
8 Stereoselective reactions
8.1 Stereochemical aspects of organic synthesis
We have studied enantiomers and diastereoisomers, their properties, methods of detection, separation etc. In this section we look at stereochemistry in the context of synthesis. To start with, some important definitions:
8.1.1 Stereospecific reactions
A stereospecific reaction is one which, when carried out with stereoisomeric starting materials, gives a product from one reactant that is a stereoisomer of the product from the other. 'Stereospecific' relates to the mechanism of a reaction, the best-known example being the SN2 reaction, which always proceeds with inversion of stereochemistry at the reacting centre.
Examples:
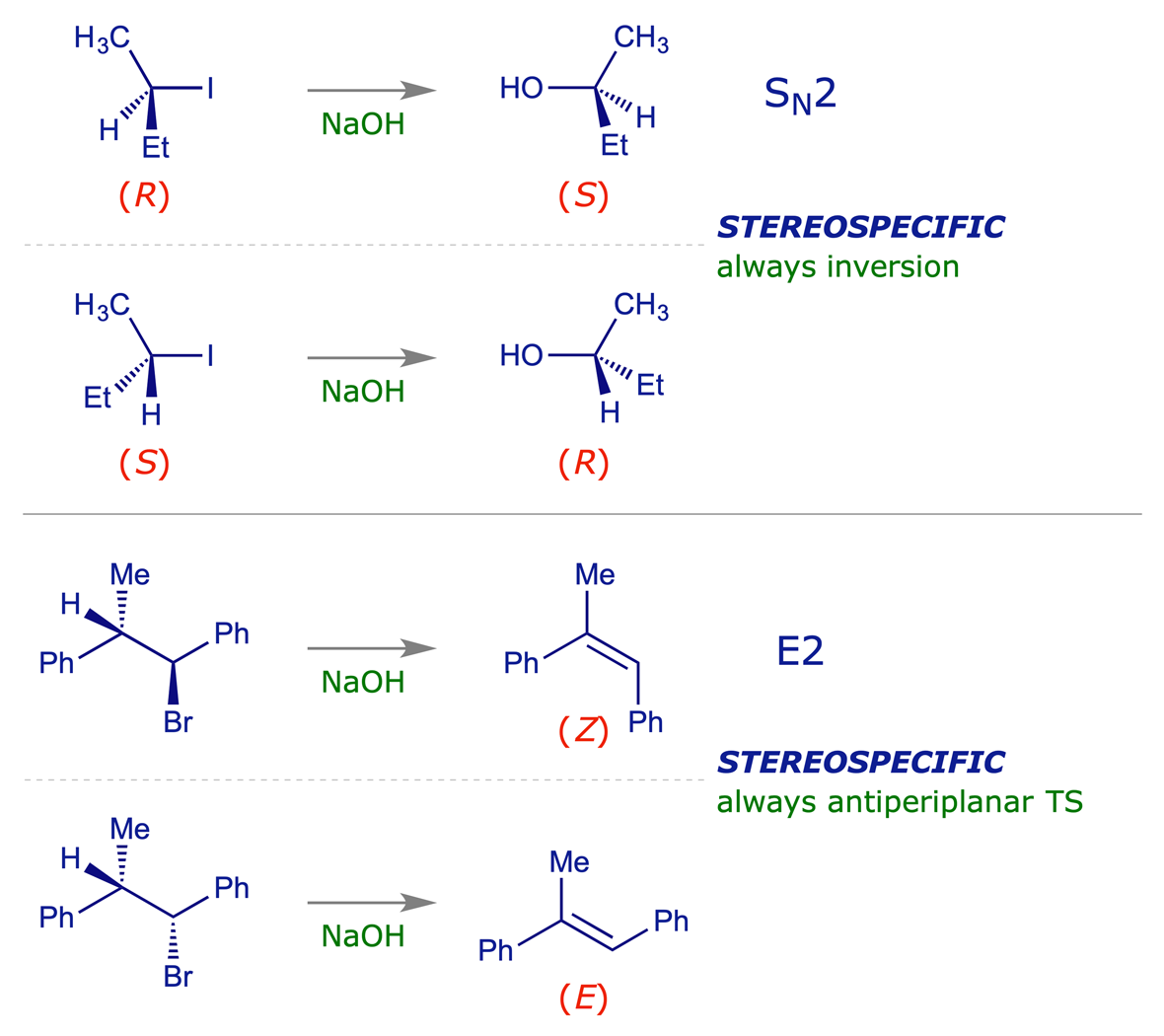
8.1.2 Stereoselective reactions
A stereoselective process is one in which one stereoisomer predominates over another when two or more may be formed. If the products are enantiomers, the reaction is enantioselective; if they are diastereoisomers, the reaction is diastereoselective. Stereoselectivity is dominated by the structural features of the reactants.
Example:

The reaction above is stereospecific (only syn addition) but the stereoselectivity is low (ca. 2:1). To understand such a reaction we must analyse it mechanistically. We will then also see that optically active products cannot be created using achiral (or racemic) starting materials in an achiral solvent. The product in such a case must also be achiral (or racemic).

8.2 The mechanistic basis of stereoselectivity
In this brief analysis we can only scratch the surface of organic synthesis and look at just a few illustrative examples of selectivity. To be of synthetic use, a reaction must be reliable, predictable and selective – features which must be analysed from a mechanistic viewpoint.
The chemistry of alkenes, alkanols and carbonyl compounds provides the core of organic synthesis. Addition reactions of double bonds (C=C or C=O) can easily provide us with new stereogenic centres, and we need to know how to exploit them.
8.2.1 Addition of X2 to alkenes
There is no regiochemical issue in the addition of a halogen (Br2, Cl2 etc.) to an alkene because the E and Nu are the same, but the question of stereoselection remains mechanistically significant. In the addition of HX to an alkene we saw that the addition of the electrophile (H+) leads to the build-up of positive charge on the adjacent carbon, eventually giving rise to a planar carbocation. Subsequent reaction with a nucleophile can in principle take place on either face of this cation, which produces a mixture of syn- and anti-addition products. In contrast, the bromination of simple alkenes is stereospecific; it is anti-selective due to the intermediate formation of a cyclic bromonium ion.
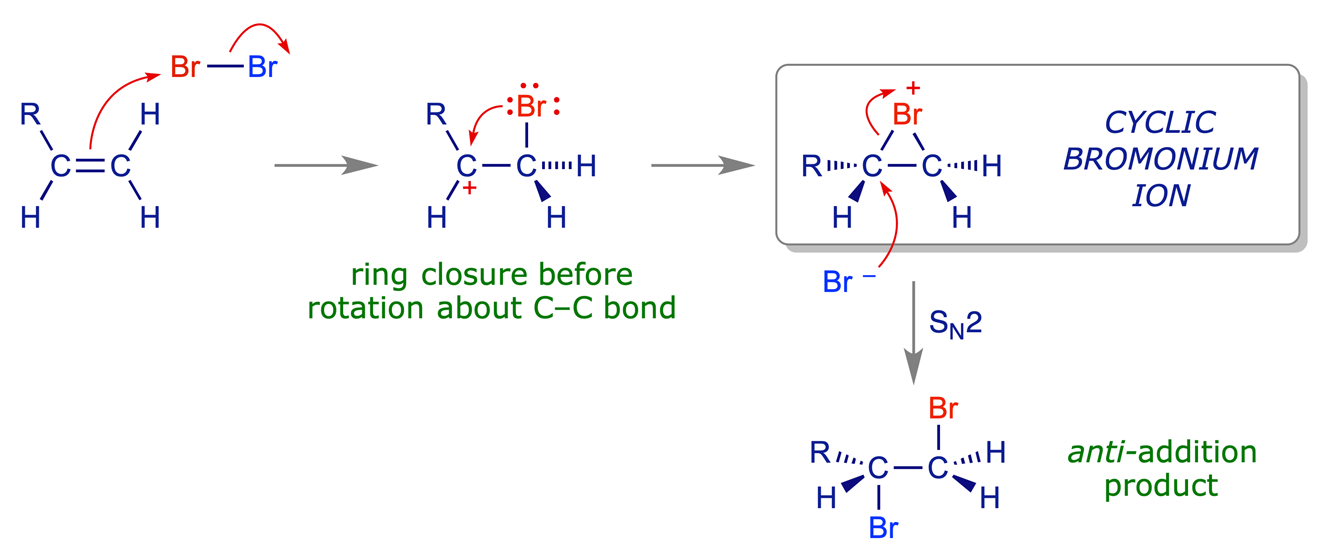
The overall process is anti-addition of Br2 to the double bond because the cyclic bromonium ion undergoes ring-opening in an SN2 process (i.e. strictly by inversion) in which the Br− counterion serves as the nucleophile. This SN2 step takes places at the most electropositive carbon of the cyclic ion (i.e. the location of the original positive charge).

The attacking nucleophile can also be the solvent, and the resulting combination of versatility and stereospecificity makes the reaction very useful in synthesis. Chloronium ions have also been observed but they are much more reactive as electrophiles (for example, they react with benzene). The tendency for bridging is F < Cl < Br < I.
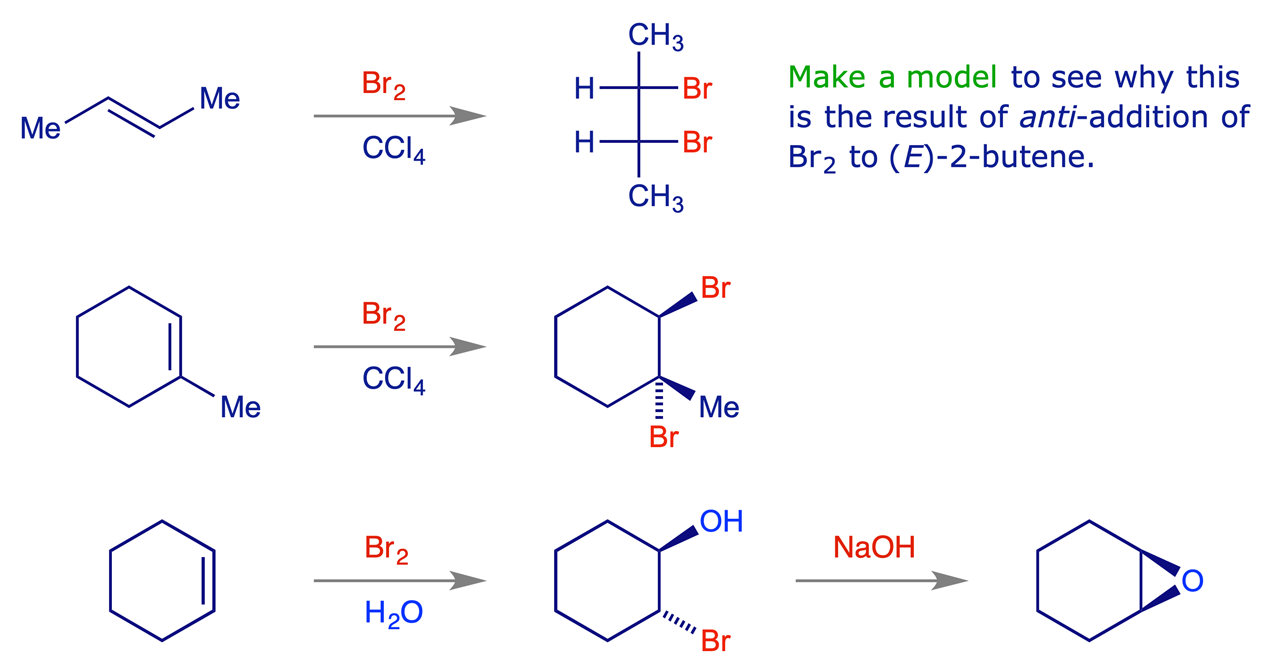
When the cationic centre is strongly stabilised by a structural feature, e.g. an aromatic ring (resonance-stabilised cation), the non-selective mechanism via a free carbocation competes with the bridged ion pathway, and some syn-product is also formed.
Example:

8.3 Generating stereogenic centres with achiral substrates
We have just seen a reaction in which stereogenic centres are generated from planar carbon. Carbonyl (C=O) and alkene (C=C) bonds are prochiral and their chemistry provides many examples which illustrate the principles of stereoselectivity in synthesis.
8.3.1 Nucleophilic addition to prochiral carbonyl groups
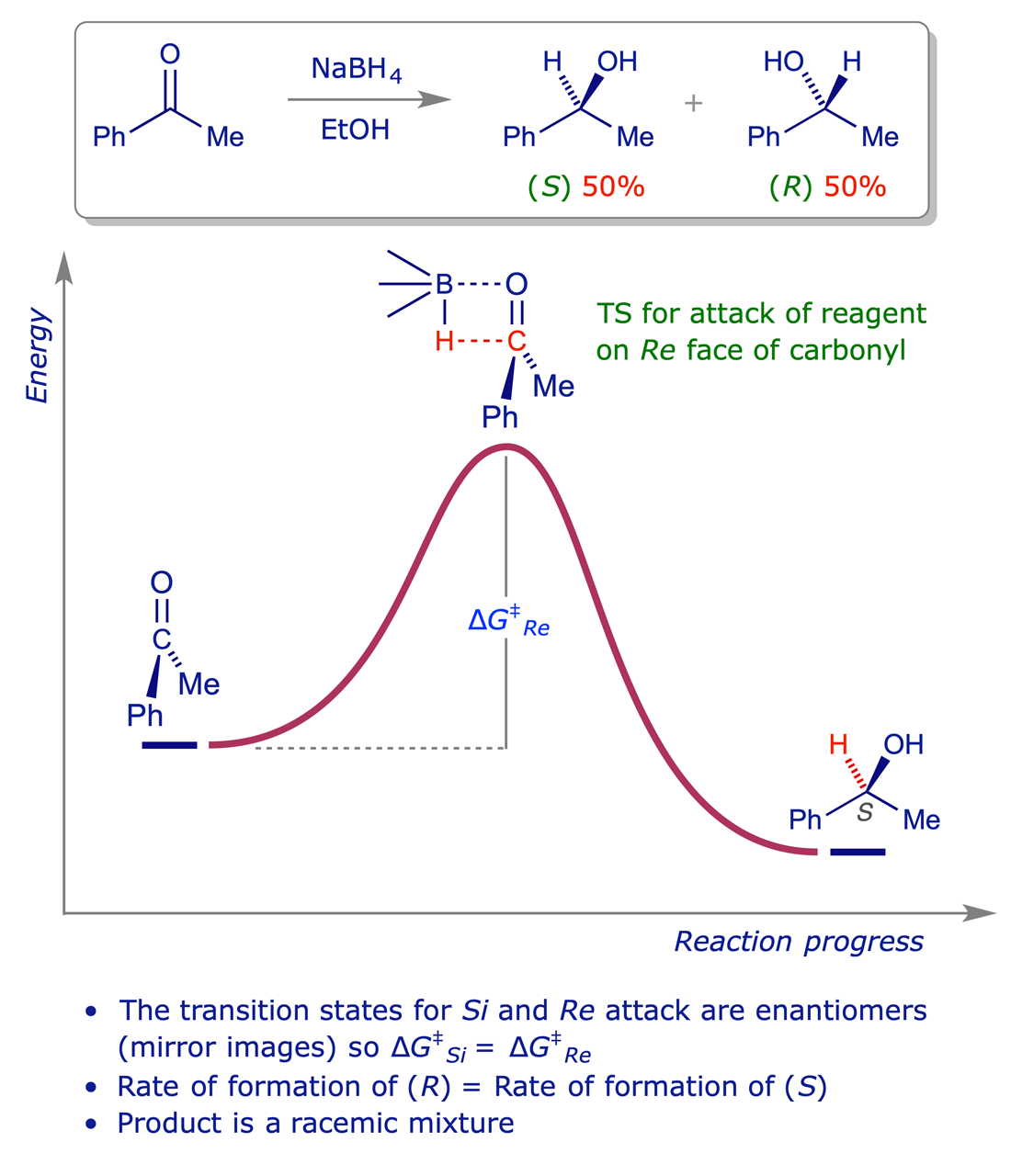
The reduction of acetophenone, which is achiral, with sodium borohydride gives 1-phenylethanol, which contains a stereogenic centre. However the product is racemic, and we must analyse the mechanism to understand why this should be the case. Two transition states are possible for bonding between the achiral BH4− and the achiral (planar) ketone, one leading to the (S)-alcohol and the other to the (R)-alcohol. But the two transition states are enantiomeric, and must therefore have identical physical properties. The activation barrier (ΔG‡) leading to each of them is the same, so they form at exactly the same rate (cf. the Arrhenius equation).
8.3.2 Hydroboration of alkenes
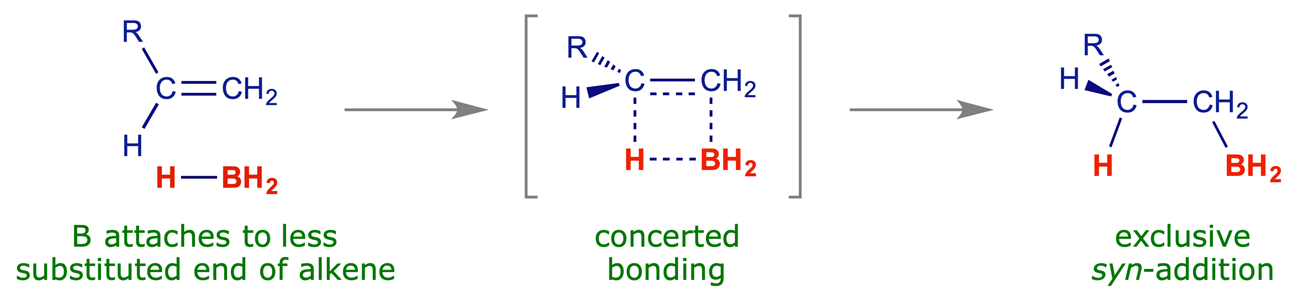
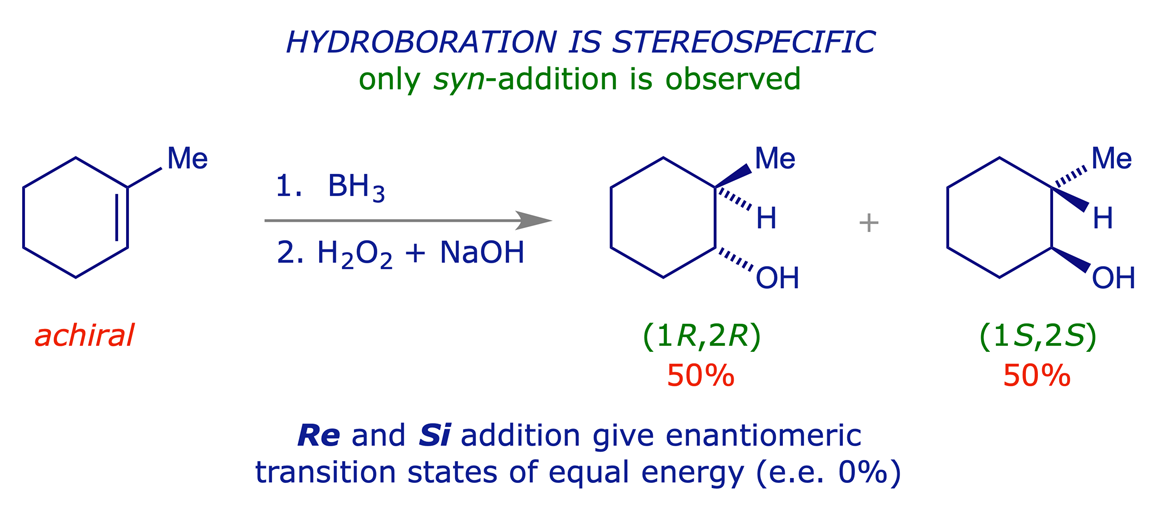
Hydroboration is an important synthetic process. Borane is electron-deficient and readily reacts with π-bonds (which are electron-rich). The addition is regioselective – the boron atom adds to the least substituted end of the double bond. The addition is stereospecific, with the B and H atoms always adding to the same face of the double bond (syn-addition).
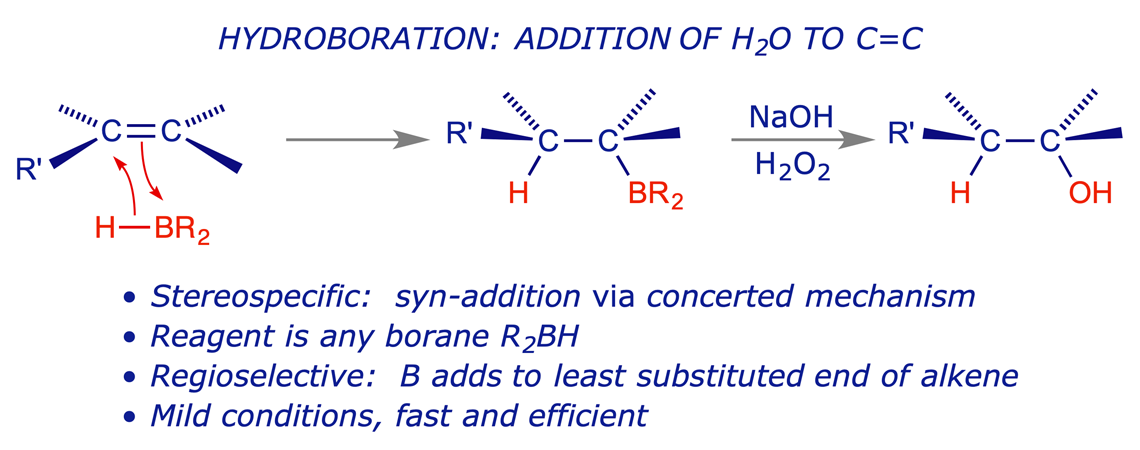
The full mechanism of hydroboration is shown in Appendix B (PDF, opens in new window). The first step is the concerted regioselective addition of the alkene π-bond (electron rich) to a B–H bond (electron poor):
With an achiral alkene the addition can proceed with equal facility on the Re and Si faces of the double bond, through enantiomeric transition states, leading to a chiral but racemic product.
8.4 Generating stereogenic centres with chiral substrates
When a starting material already possessing a stereogenic centre undergoes a reaction which leads to the generation of a new one, diastereoselectivity (and how to control it) becomes a major consideration.
8.4.1 Nucleophilic addition to a homochiral cyclic ketone
Whereas the addition of a nucleophile to the two faces of acetophenone (Section 8.3.1) leads to a pair of enantiomers, the prior presence of the stereogenic centre in 2-methylcyclopentanone means that the two faces of the carbonyl group are diastereotopic (rather than enantiotopic), and hydride addition can lead to two separable products (diastereoisomers) in unequal amounts. In this reaction the approach of the hydride reagent to the C=O group is easier from the rear (Si) face, further away from the large methyl group. Our starting material is enantiomerically pure (2S)-enantiomer, so each of the two diastereoisomers produced will also be enantiomerically pure.

Addition reactions like this are irreversible and so proceed under kinetic control, the product ratio reflecting the relative rates of the two modes of addition. These are different because the two transition states are diastereoisomeric and have unequal energies. Steric effects in the transition states determine their energies and hence the product ratio. This is a typical example of steric approach control, in which a substrate reacts preferentially at the least hindered site. The more bulky the reagent, the bigger the preference for attack from the least hindered face.
The proximity of the 2-methyl group to the prochiral reaction site (C-1) engenders a high level of steric approach control in the above reaction, and the cyclic (inflexible) nature of the ketone ensures that the methyl group cannot get away from the bond-forming process. More distant stereogenic centres would be expected to exert less influence on reactions at a prochiral sites.

8.4.2 Nucleophilic addition to a racemic cyclic ketone
To remind yourself of the fundamental principle that non-racemic products cannot be created from racemic starting materials in an achiral medium, consider what would happen if the reaction in the previous section was performed on racemic 2-methylcyclopentanone. The outcome is shown below.

The ratio of diastereoisomers formed in the reaction is 76:24 (i.e. the d.e. is 52%), and this will be the same for both enantiomers of the starting material. So we can say:
- Racemic ketone gives 76% yield of racemic cis-2-methylcyclopentanol
- If the e.e. of the starting ketone is 60%, then each product will each have an e.e. of 60%
8.4.3 Enantioselective hydride reduction of carbonyl compounds
The reduction of acetophenone with NaBH4 (section 8.2.1) confirmed that achiral hydride reducing agents cannot react in an enantioselective manner. However, Yamaguchi and Mosher found that the partial decomposition of lithium aluminium hydride (LiAlHa) with a chiral alcohol gave a modified chiral reducing agent capable of reducing acetophenone enantioselectively. Although the method is simple, it should be noted that the level of enantioselection (e.e. 68%) is not ideal in a practical sense because of the difficulties associated with purifying (i.e. resolving) the product in order to obtain a single enantiomer.

Reference: S. Yamaguchi and H. S. Mosher, J. Org. Chem., 1973, 38, 1870–1877 (https://doi.org/10.1021/jo00950a020).