1 Isomerism
1.1 Types of isomerism
Isomers are broadly defined as different compounds which have the same molecular formula. There are several ways in which compounds with the same molecular formula can be isomeric, but they all fall into one of two subclasses: constitutional isomers or stereoisomers.
1.1.1 Constitutional isomers (structural isomers)
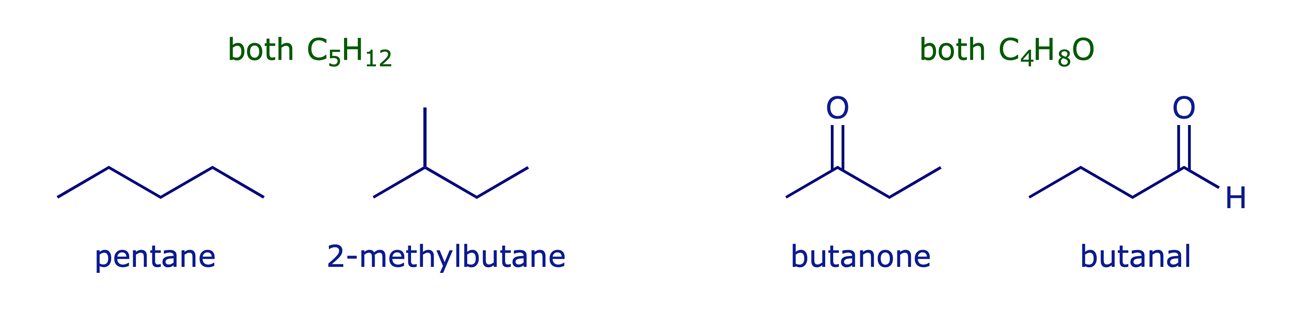
Constitutional isomers have the same molecular formula but different connectivity between atoms. They may have similar properties, but essentially they are different compounds.
Examples:
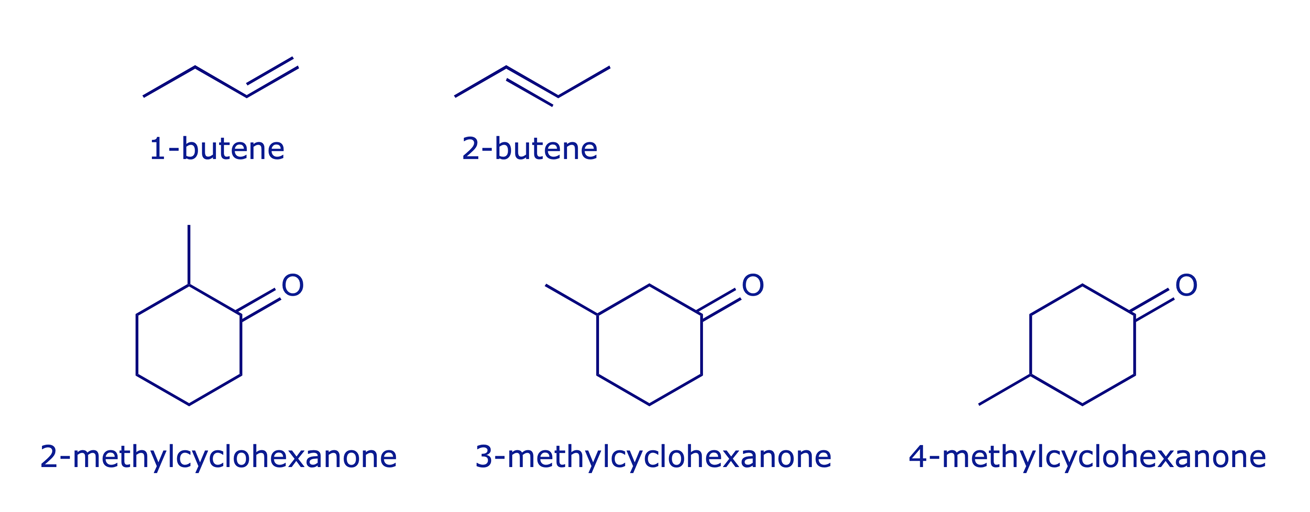
Regioisomers are a class of constitutional isomers which have the same functional groups but attached at different positions.
Examples:
1.1.2 Stereoisomers
Stereoisomers have the same constitution (atom-bond connectivity) but the atoms are oriented differently in space. Stereoisomers can be subdivided into two classes.
1.1.2.1 Conformational isomers
These can be interconverted by free rotation about single bonds. The interconversion of conformations is usually rapid at room temperature because the energy barrier to rotation is low, so these isomers normally cannot be separated from one another.
1.1.2.2 Configurational isomers
Configurational isomers differ in the spatial arrangement of their atoms and can only be interconverted by breaking bonds (and not by free rotation about single bonds). In the case of carbon compounds they do not interconvert at room temperature and can be separated using physical methods.
Examples:

Configurational isomers fall into two categories: enantiomers and diastereoisomers. We will start by analysing some fundamental structural issues involving conformational isomerism and the structures of cyclic alkanes.
1.2 Conformations of acyclic alkanes
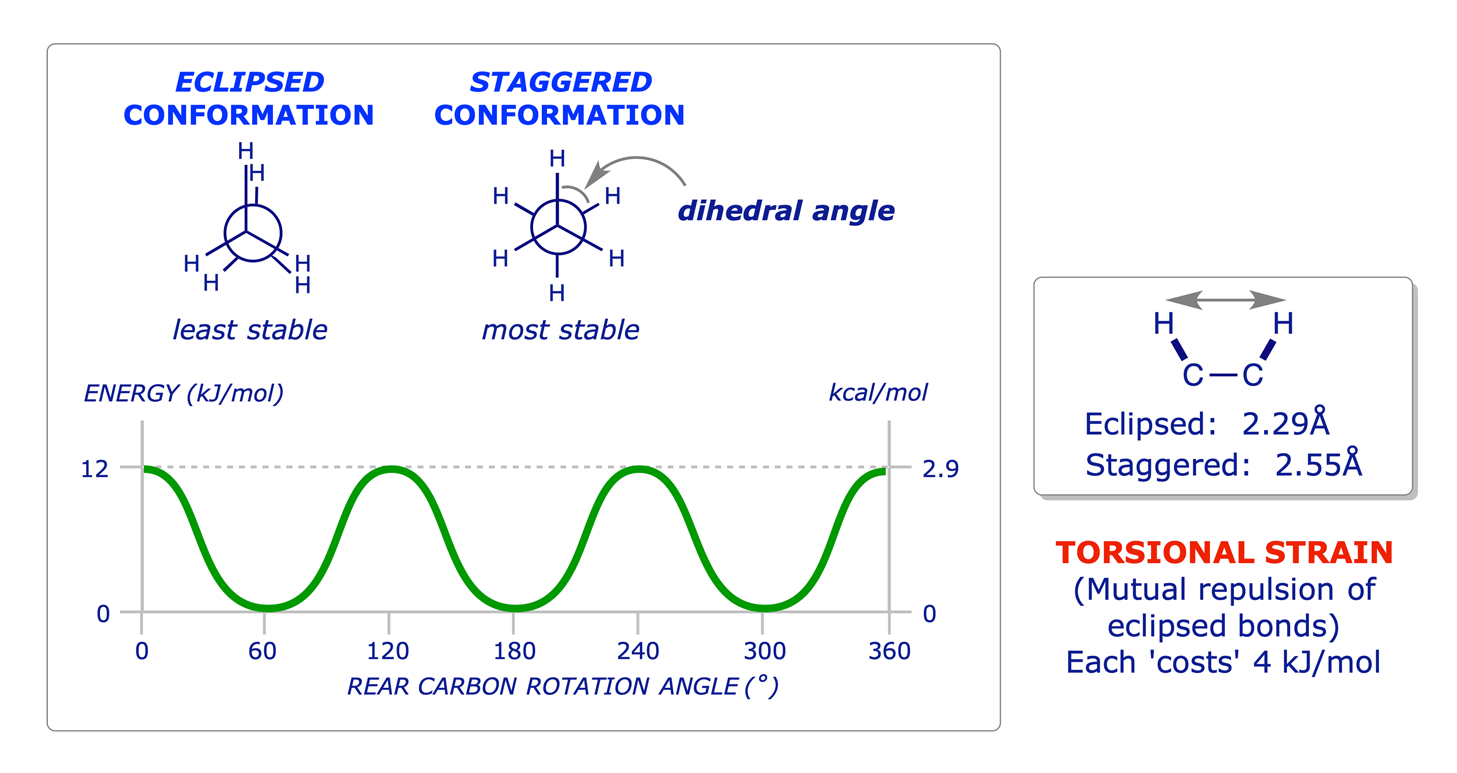
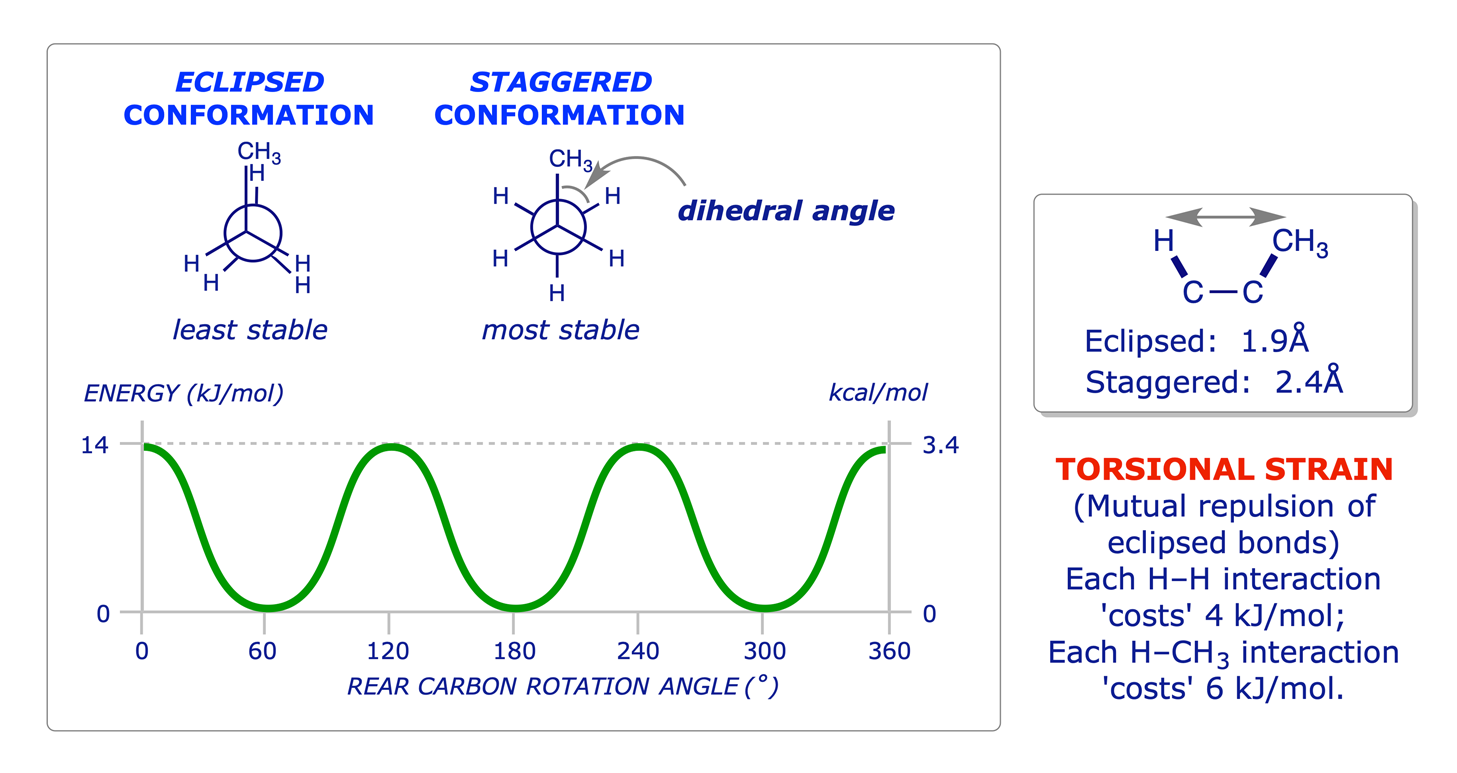
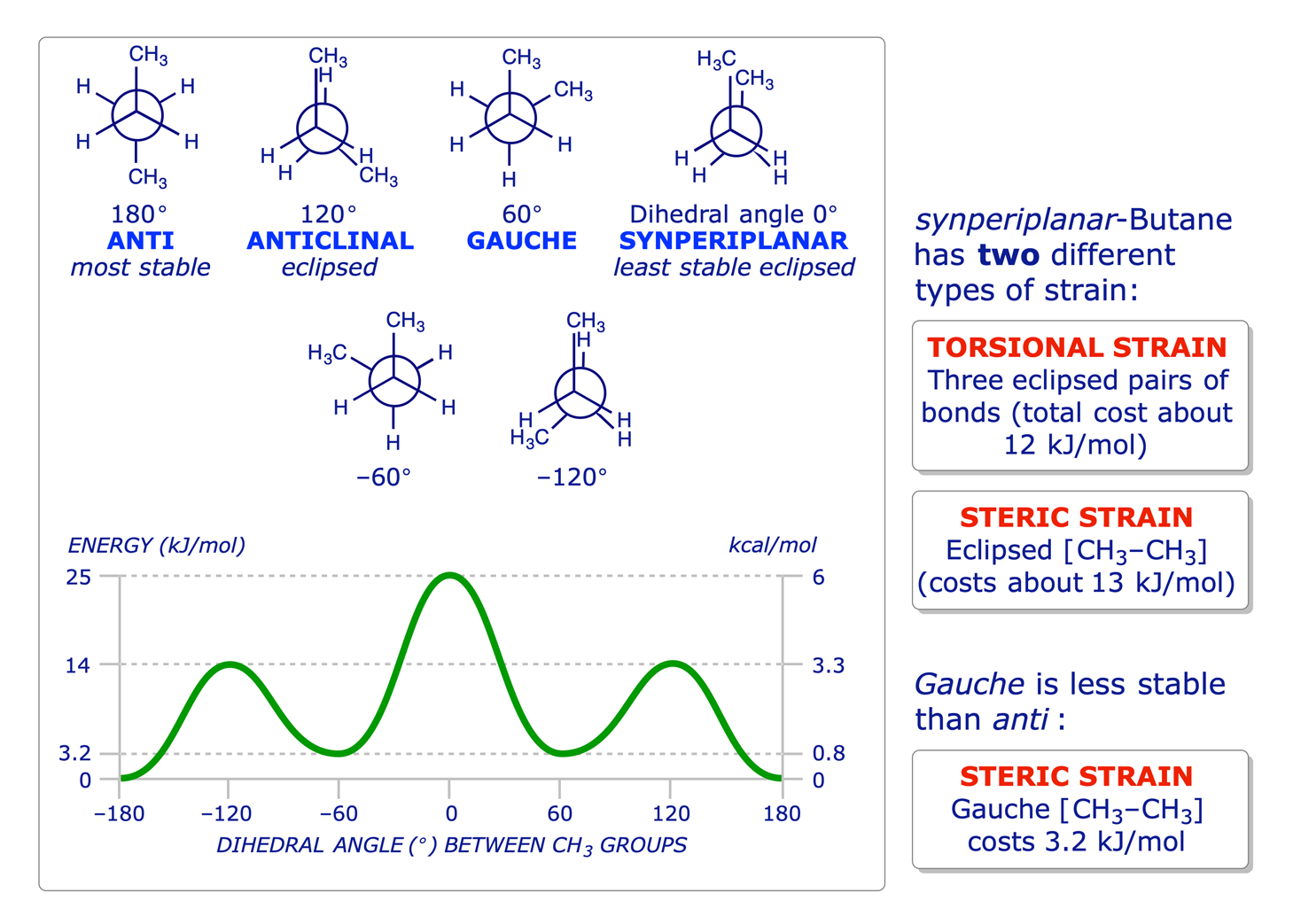
The principles of conformation were introduced in the Level 1 course with reference to ethane and propane. The use of Newman projections was introduced to facilitate the study of this topic, and it is known that staggered conformations are more stable than eclipsed conformations because of two different types of strain: torsional and steric.
1.2.1 Ethane
1.2.2 Propane
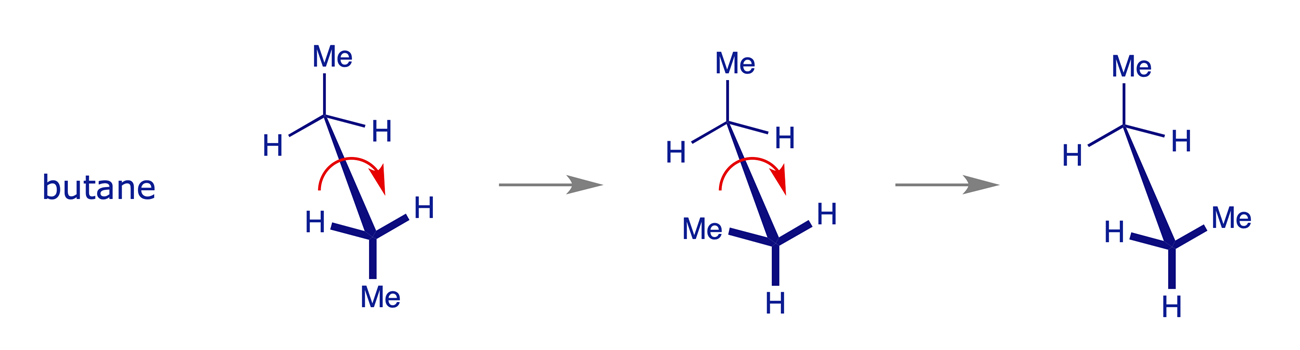
1.2.3 Butane
1.2.4 Higher alkanes
The principles illustrated above for butane also apply to pentane, hexane and the higher alkanes. The most favorable conformation for any alkane is the one in which the carbon-carbon bonds have staggered arrangements and in which large substituents are arranged anti to each other. A generalised alkane structure is shown below.
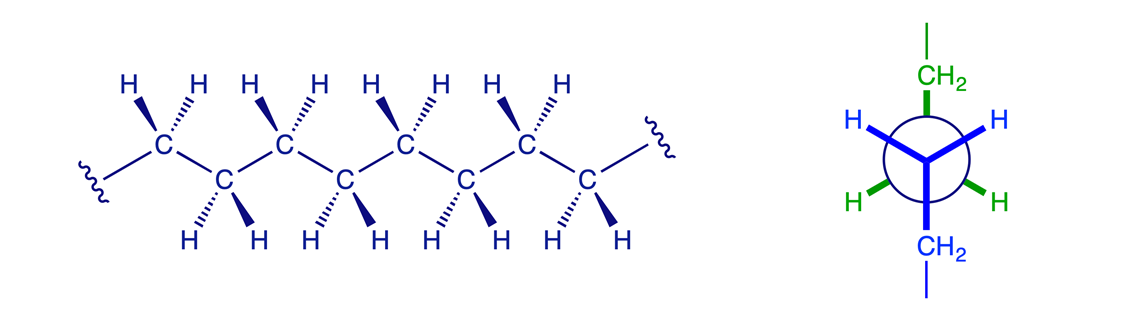
At room temperature, enough thermal energy is present to cause rotation around sigma bonds to occur rapidly so that all conformers are in equilibrium. At any given instant, however, a larger percentage of molecules will be found in a more stable conformation than in a less stable one.
1.3 Cyclic alkanes
In a paper published in 1885 the German chemist Adolf von Baeyer proposed that the cyclic alkanes were planar. His opinion was based on his observations that, with the exception of cyclopentane and cyclohexane, cyclic alkane rings were (i) rarely encountered in nature and (ii) difficult to synthesise. If cycloalkanes were planar, von Baeyer argued, only those whose bond angles were close to the tetrahedral angle (109.5°) would be stable. The others would have angle strain arising from the distorted bond angles at the carbon atoms.
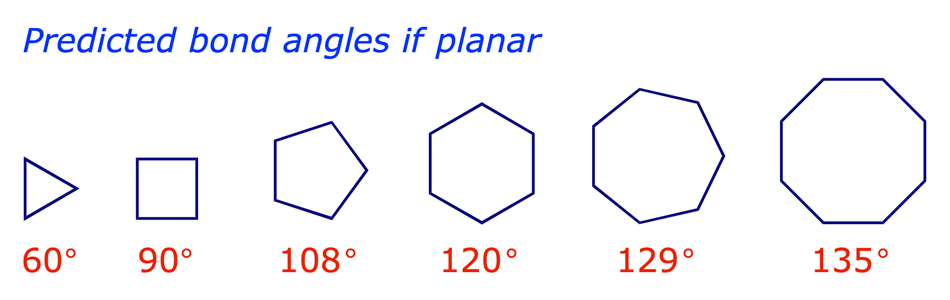
Heats of combustion measurements later showed that the Baeyer strain theory was wrong for the large cycloalkanes, but confirmed the angle strain in cyclopropane and cyclobutane.
1.3.1 Cyclohexane
It was eventually proved that cyclohexane is not planar but puckered in a 3-D conformation that relieves strain. Its most stable arrangement is referred to as the chair conformation, in which every C–C–C bond angle is 111° (very close to the tetrahedral angle) and every adjacent pair of C-atoms has staggered bonds.
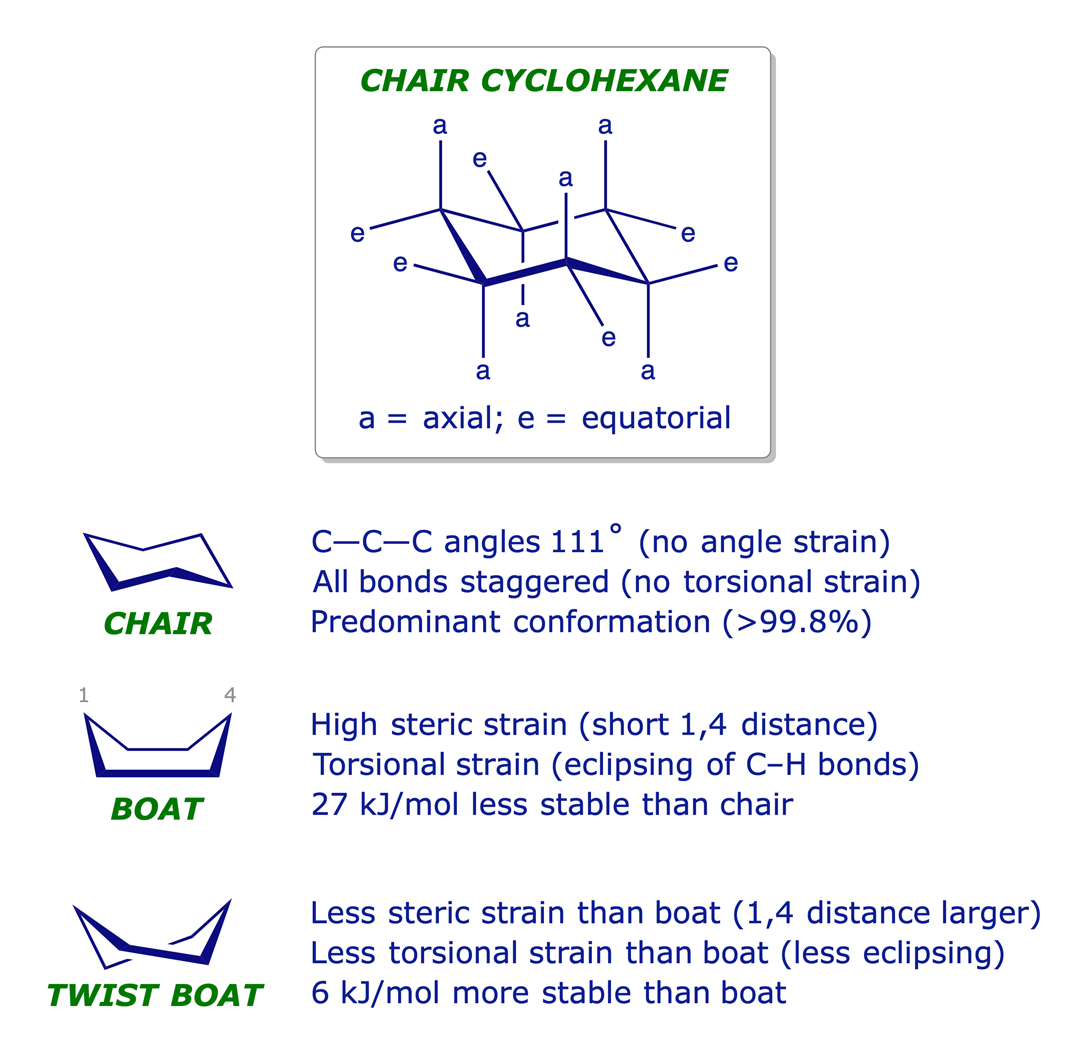
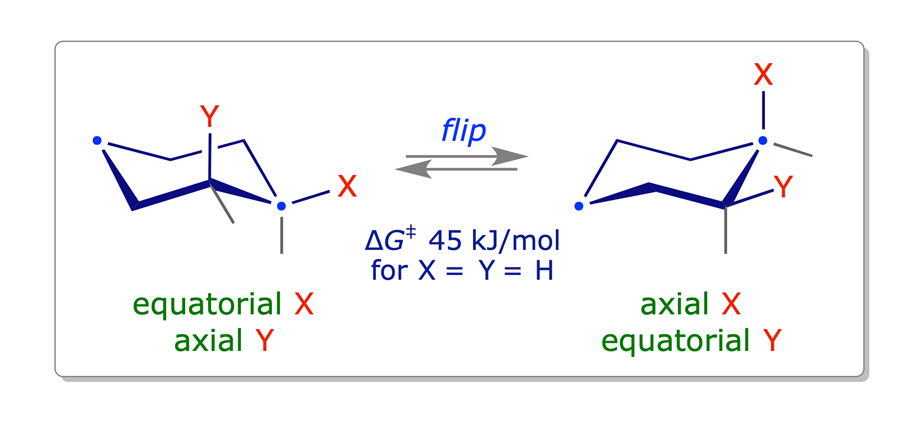
The different chair conformations of cyclohexane can interconvert or 'flip' very easily: the activation barrier is low — about 45 kJ/mol.
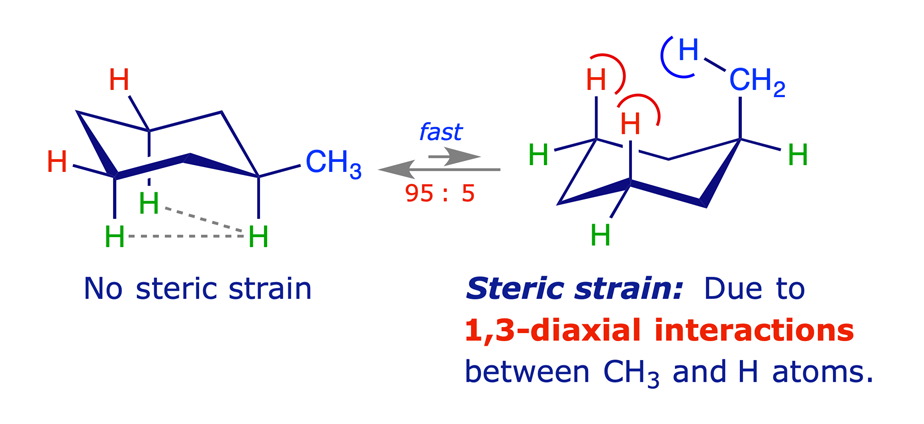
1.3.2 Methylcyclohexane
Equilibration (flipping) of the ring in methylcyclohexane gives rise to two chair conformations with different energies. The more stable of these has the methyl group equatorial, thereby avoiding the steric strain arising from 1,3-diaxial (gauche) interactions. Other monosubstituted cyclohexanes behave similarly, the larger the substituent the higher percentage of the equatorial conformer in the equilibrium mixture.
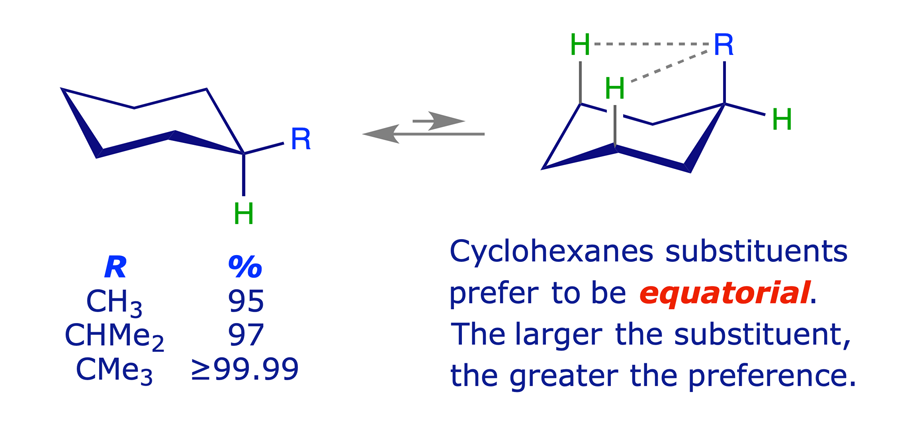
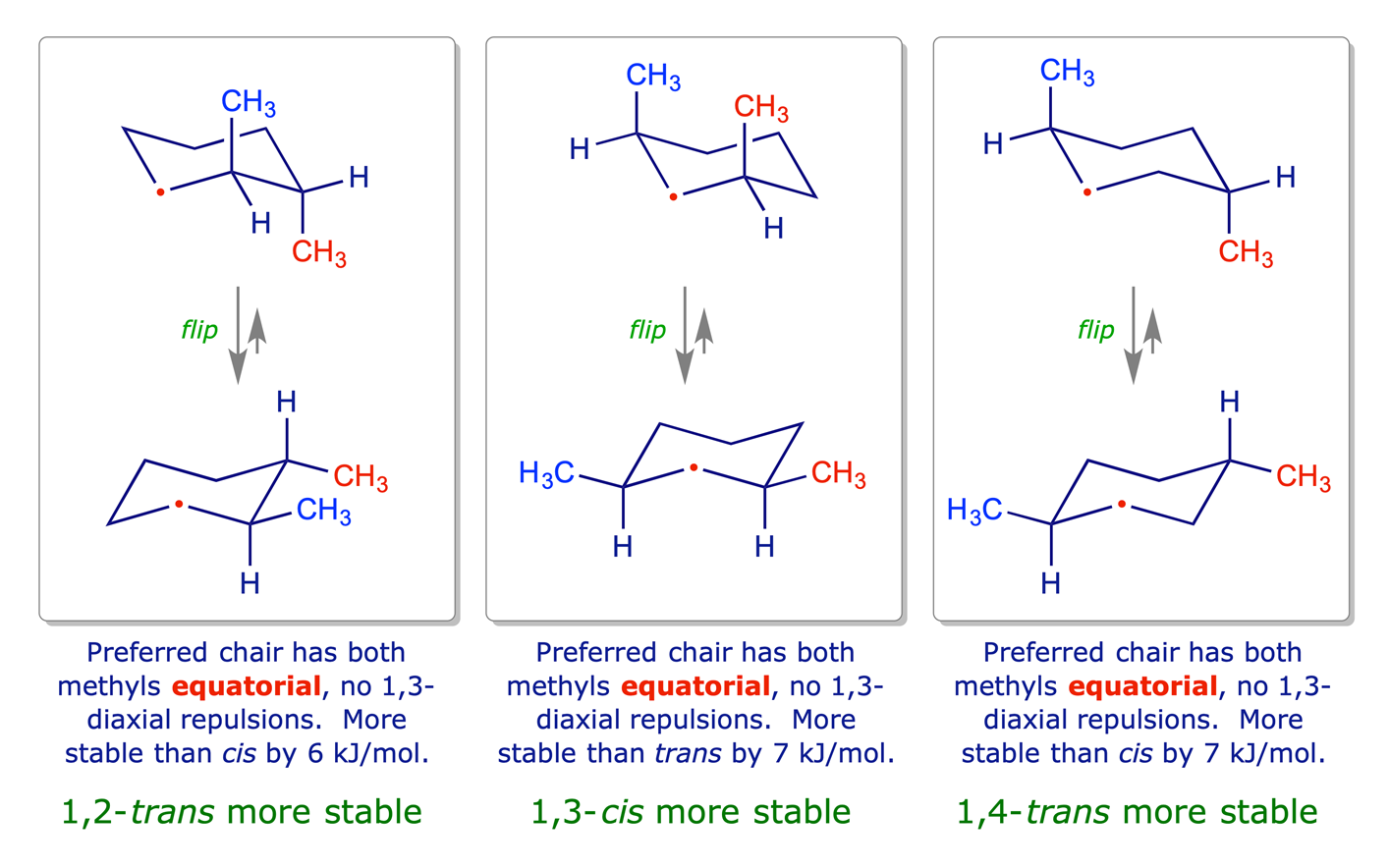
1.3.3 Disubstituted cyclohexanes
In disubstituted systems the conformational (axial/equatorial) preference depends on the substitution pattern. The lower energy arrangement has the larger of the two groups in an equatorial position, so for X = Y in the graphic below, the all-equatorial isomers are the more stable of each pair.
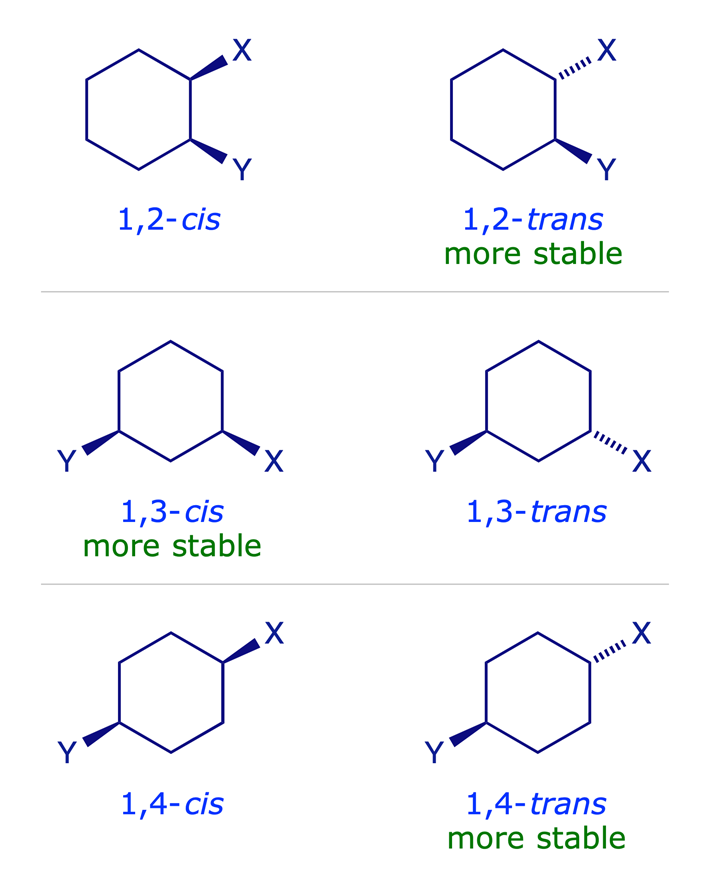
The influential factors are:
- Cyclohexane ring has chair conformation.
- Substituents adopt equatorial orientation to minimise 1,3-diaxial steric repulsions.
- For X = Y, the all-equatorial isomers are the more stable of each pair.
- For X ≠ Y, the preference will be for the larger of the two groups X and Y to be equatorial.