The Role of Receptors
- Introduction
- Receptor activation
- Receptor mechanisms
- Cimetidine (Tagamet®) and Ranitidine (Zantac®)
- Ion channel receptors
- G-Protein-coupled receptors
- Kinase-linked receptors
- The development of imatinib (Gleevec®)
1 Introduction
Receptors are the most important drug targets in medicine. They are the proteins that mediate signalling and response, and are therefore implicated in just about every type of condition and the body's response to it. This is a global topic: we will focus on the underlying principles and chemistry that may be involved at the molecular level.
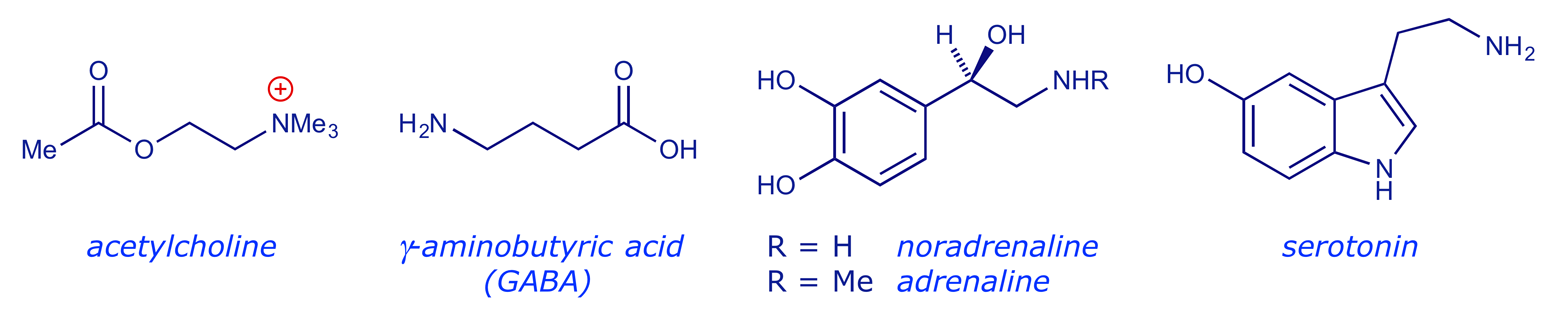
In a complex organism, all systems must interact in concert. For example, all heart muscle cells contract at the same time – issues of supply and demand are finely coordinated, and the central nervous system (CNS) provides the necessary control via the brain and spinal column. But the CNS does not connect directly to cells — the signal is carried across a gap by a neurotransmitter molecule. On reaching a cell surface, the messenger molecule binds and interacts with a specific protein (receptor) embedded in the cell membrane. This event produces a biological effect inside the cell, e.g. the contraction of a muscle cell or the activation of a biosynthetic pathway.

A variety of molecules can interact with biological receptors. Some are very simple (Ca2+ can act as a chemical messenger) while others are more complex (cf. angiotensin II). A messenger molecule may operate over a short distance (the nerve signal in the above figure jumps a gap of about 100 Å) or may have a long distance to travel — hormones such as adrenaline are released in one part of the body and travel in the bloodstream to their target receptor. But however far they travel, their action is the same. They interact with a receptor, the message is received by the cell, and the cell adjusts internally, i.e. there is a biological response. One way to control the biological state of the cell is to tap into its signalling processes.
2 Receptor activation
There are three different families of membrane-bound receptor (ion channel; G-protein-coupled; kinase-linked). Receptor 'switching' occurs through binding of a messenger molecule to the extra-cellular part of the receptor, eliciting an effect on the intracellular part.
A receptor protein is usually embedded within a cell membrane, with part of its structure on the outside of the cell. As with an enzyme, the receptor protein's tertiary structure gives rise to a potentially complex 3-D shape which is subject to a variety of intra- and intermolecular forces. This 3-D shape will include an area — the binding site — that can receive the smaller 'messenger' molecule, when it arrives, by a process of induced fit. The presence of the messenger causes the receptor to 'switch on' and send its message. The induced fit principle is the same as the docking of a substrate into the active site of an enzyme, except that, with a receptor, no chemical reaction is induced. It is the slight change in the tertiary structure of the receptor, due to the presence of the messenger molecule, that opens the signalling pathway.
To understand how this might happen, we should first remind ourselves of the forces that can be involved in ligand–protein binding. These are the same whether the 'ligand' is the substrate of an enzyme, the messenger for a receptor, or a foreign molecule (i.e. drug) that we design for a particular purpose. These forces correspond to those that combine to maintain a protein's tertiary structure in the absence of any ligand:
• Hydrogen bonding
H-bonds are central in stabilising the 3-D structure of proteins and DNA (intramolecular H-bonding), as well as a means of aiding ligand–receptor binding (intermolecular H-bonding). The following list should be useful when considering the ways in which the binding of drugs to their targets might be optimised.
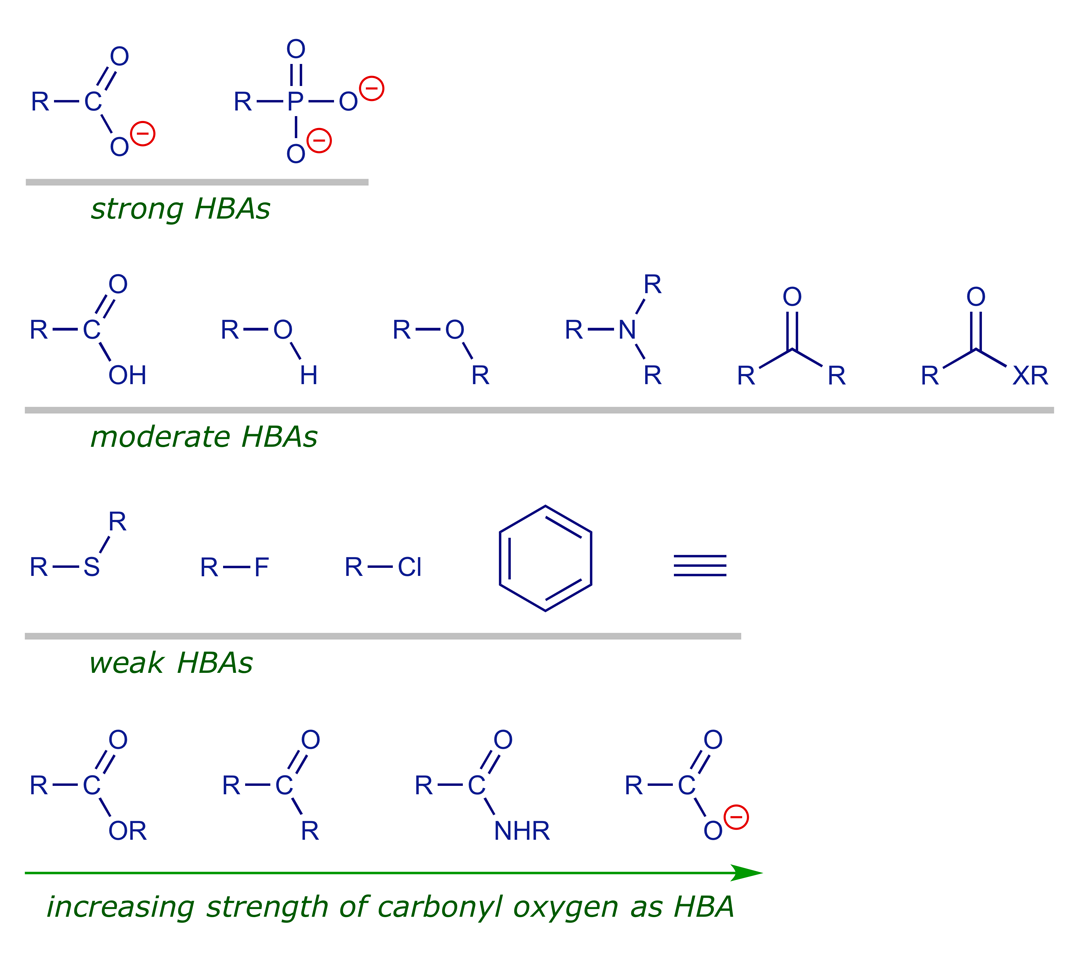
• Ion-dipole and dipole-dipole interactions
Some of the C–X bonds in the receptor and in the ligand will be polarised by virtue of the presence of electronegative atoms, commonly where X is O or N, leading to the presence of dipoles. Positively-charged sites will be attracted to negatively-charged sites (or negative charges) and vice versa.

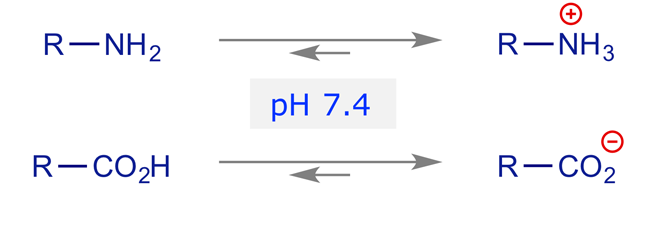
• Ionic (electrostatic) interactions
Physiological pH is generally taken to be about 7.4 and at this pH amino groups are usually protonated and carboxyl groups are usually deprotonated.
• Hydrophobic interactions
Water on its own is a complex H-bonded network of molecules, but when non-polar functions (e.g. hydrocarbons) are dissolved in water some of this H-bonding is disrupted. This makes an entropic contribution to the binding process in a phenomenon known as the hydrophobic effect.

• van der Waals interactions
Temporary dipole-dipole attractions (van der Waals forces) only become significant when there is surface contact of atoms, but under these circumstances they are important.
Since the various non-covalent interactions are relatively weak, there needs to be several co-operative binding forces before a strong interaction can take place. To illustrate, some of the possible binding interactions of 2-amino-1-phenylethanol to a receptor are shown below.
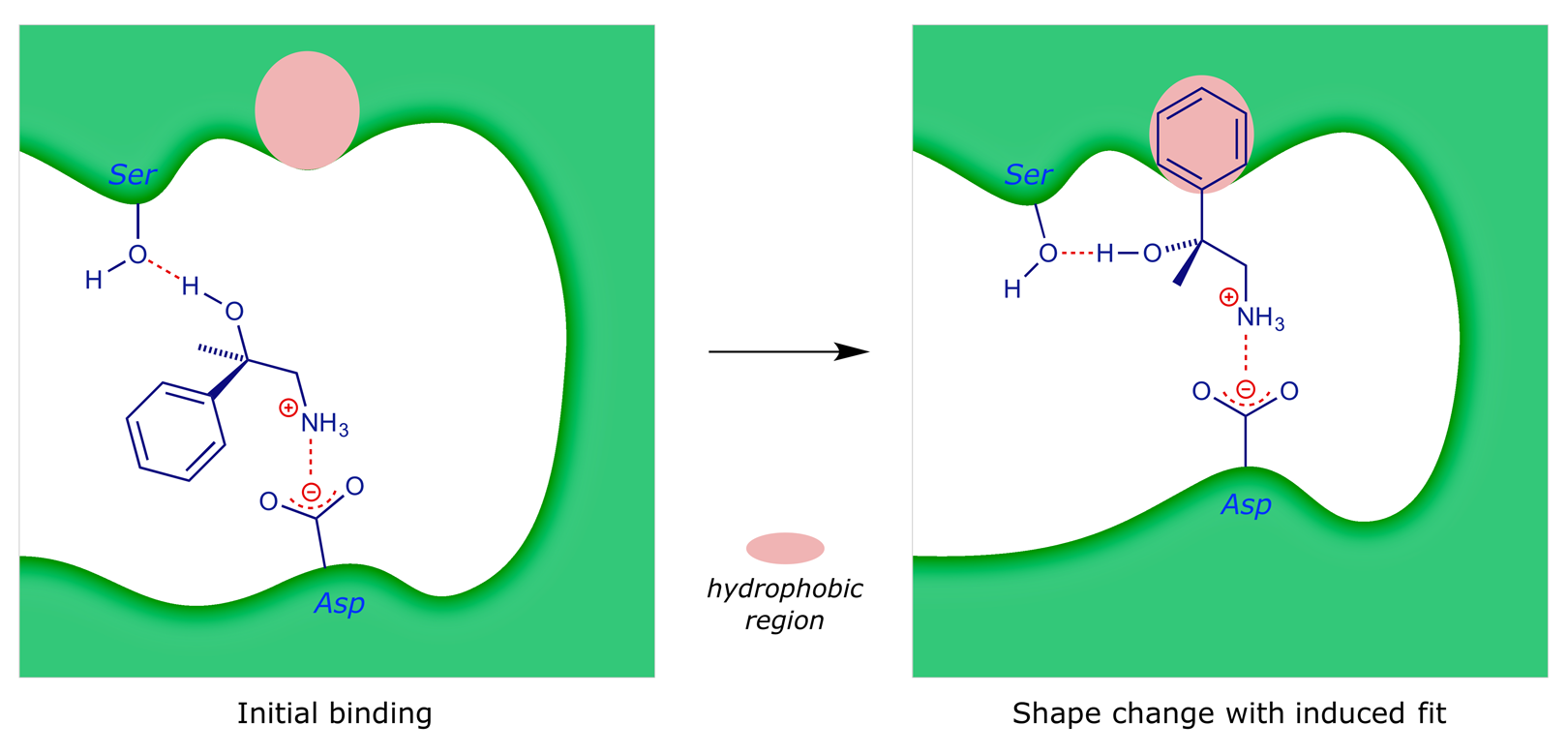
3 Receptor mechanisms
Receptors are embedded in the lipid bilayer of a cell membrane. There are three different families of membrane-bound receptor:
- ion channel receptors
- G-protein-coupled receptors 2012 Nobel Prize for Chemistry
- kinase-linked receptors
Receptor 'switching' occurs through binding of the messenger to the extra-cellular part of the receptor, eliciting an effect on the intracellular part. To see how the binding of a ligand (or drug) might affect the action of a receptor, we will consider the artificial example below.

In general a receptor needs to be embedded in the lipid bilayer in order to function: the interface that it shares with the membrane to some extent dictates its tertiary structure. Consequently any attempt to isolate and characterise a receptor protein will often lead to degradation. For this reason the precise structures of many receptors were unknown until proteomics began to provide a way to identify them without a crystal.
Our 'cartoon' diagrams represent an ion channel receptor, constructed solely for the purpose of demonstrating the principles involved in making the interaction of a protein and a ligand into a switching device. The diagrams illustrate how drugs and receptors might interact, although in the case of real receptors the binding site is generally more complex. Our receptor's extracellular surface has three binding locations.
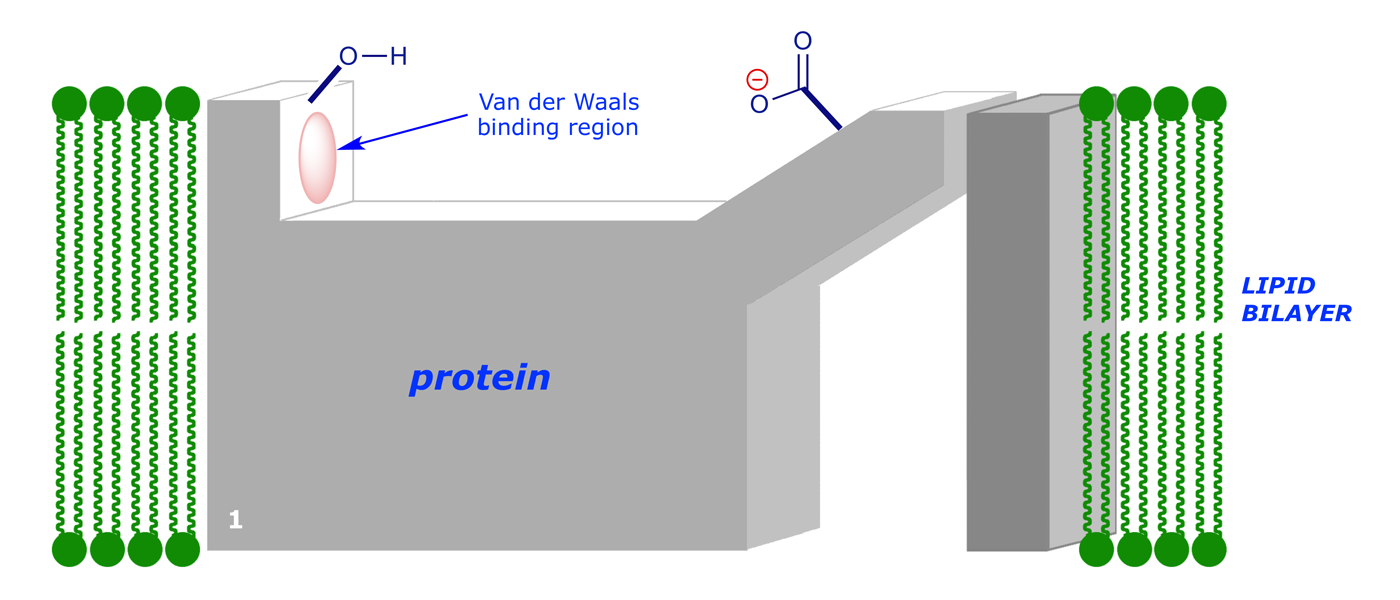
In normal operation, the signalling molecule (2-amino-1-phenylethanol) will bind to the receptor (binding areas –OH and van der Waals region) to give an initial complex:
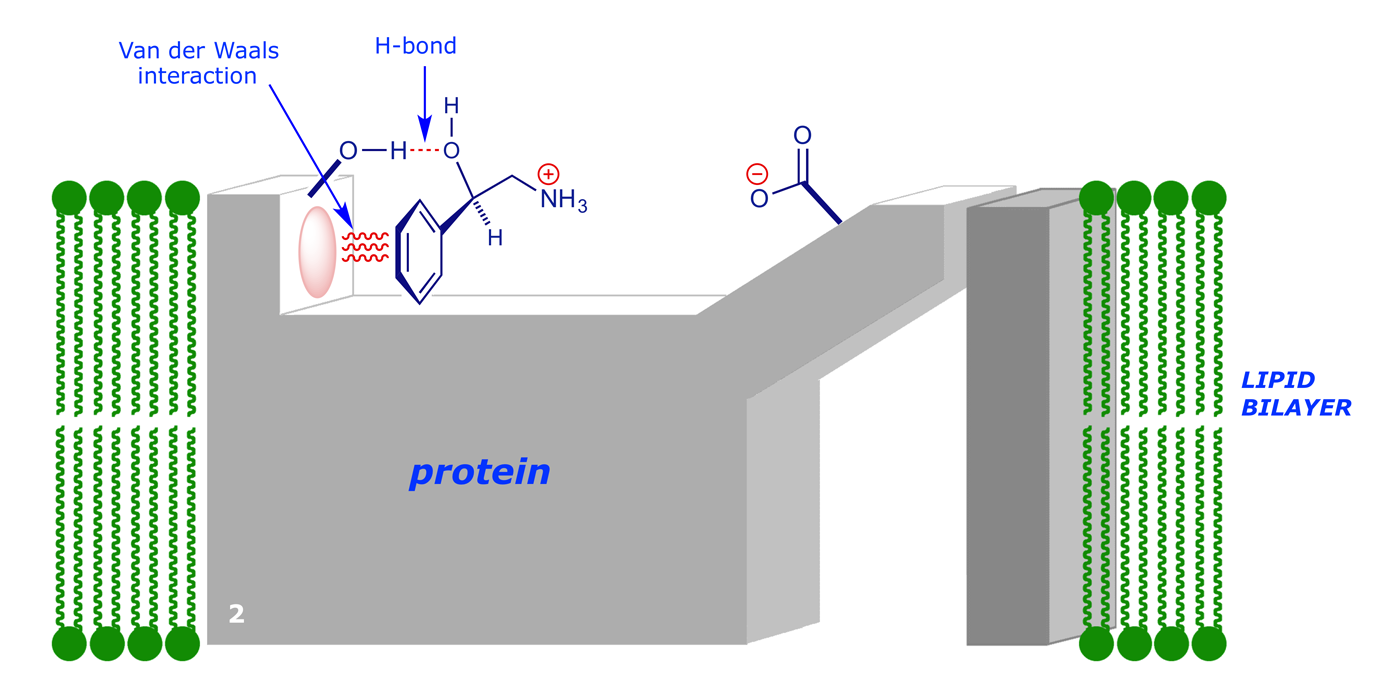
There are good van der Waals and H-bonding interactions, but the potential ionic bond with the CO2– group is not as strong as it could be, due to the distance involved. However, the complex can change its shape, i.e. undergo a conformational change, to allow tighter binding to the signalling molecule. An induced fit takes place:
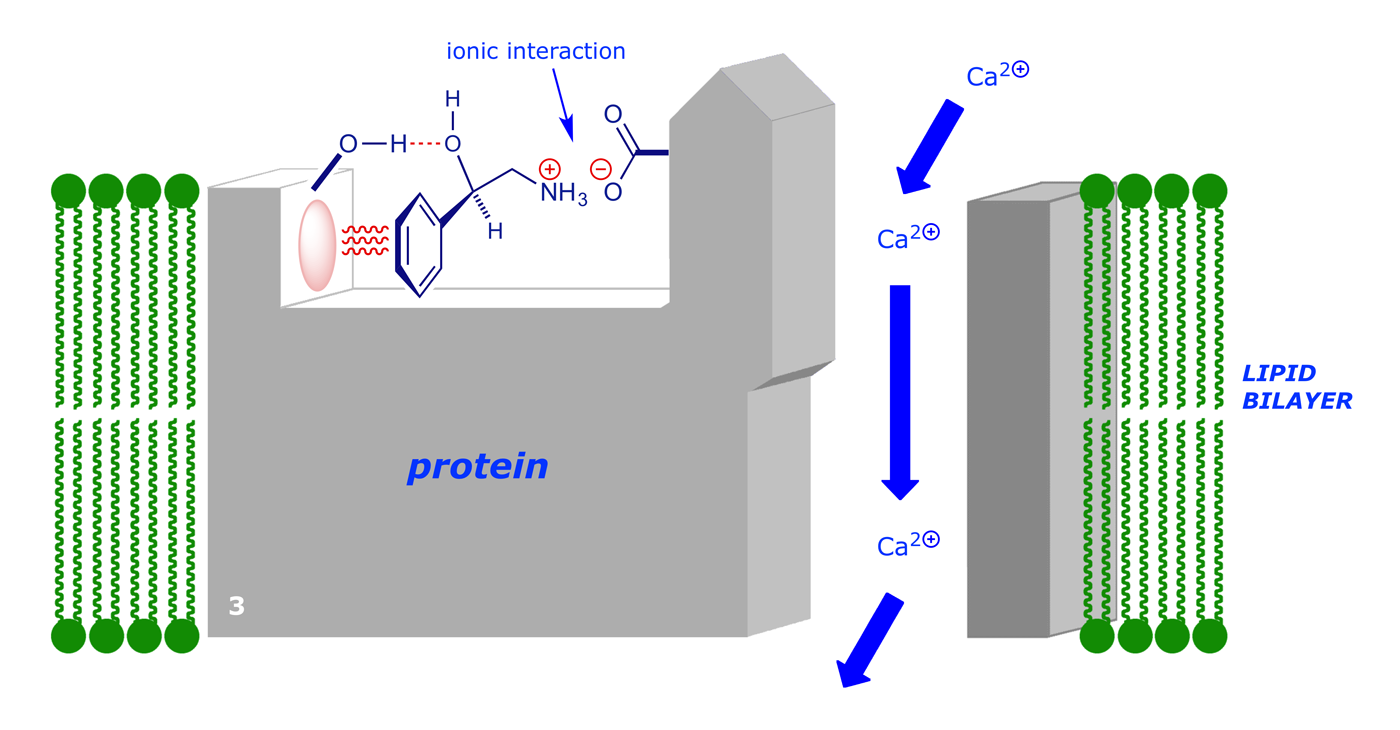
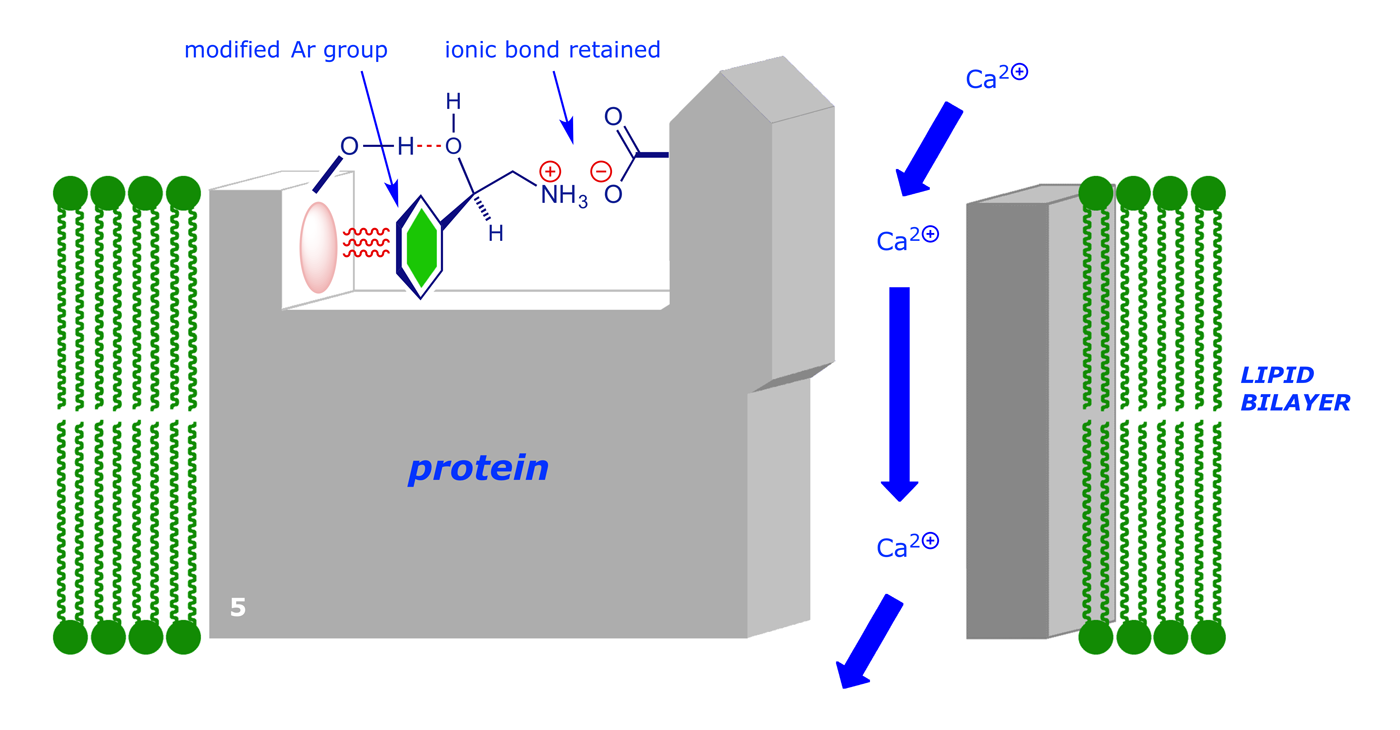
In this example, the conformational change opens up an ion channel in the cell wall and allows calcium ions to flow into the cell from the outside. This in turn triggers other effects inside the cell.
Obviously this illustration is a simplification. In reality both the messenger and the binding site take up different conformations so as to maximise their bonding interactions. It should also be recognised that the binding should not be too strong, otherwise the messenger would be unable to leave and we would not have a reversible 'switching' process. Neurotransmitters tend to leave their binding site soon after their message takes effect.
This is the normal function of the receptor, and in disease therapy we generally require to turn off this 'switch,' in this case preventing the opening of the ion channel. In trying to do this, there is no point in designing a drug to precisely mimic the natural signalling agent – if we do this the drug will open the channel (i.e. it will act as an agonist):
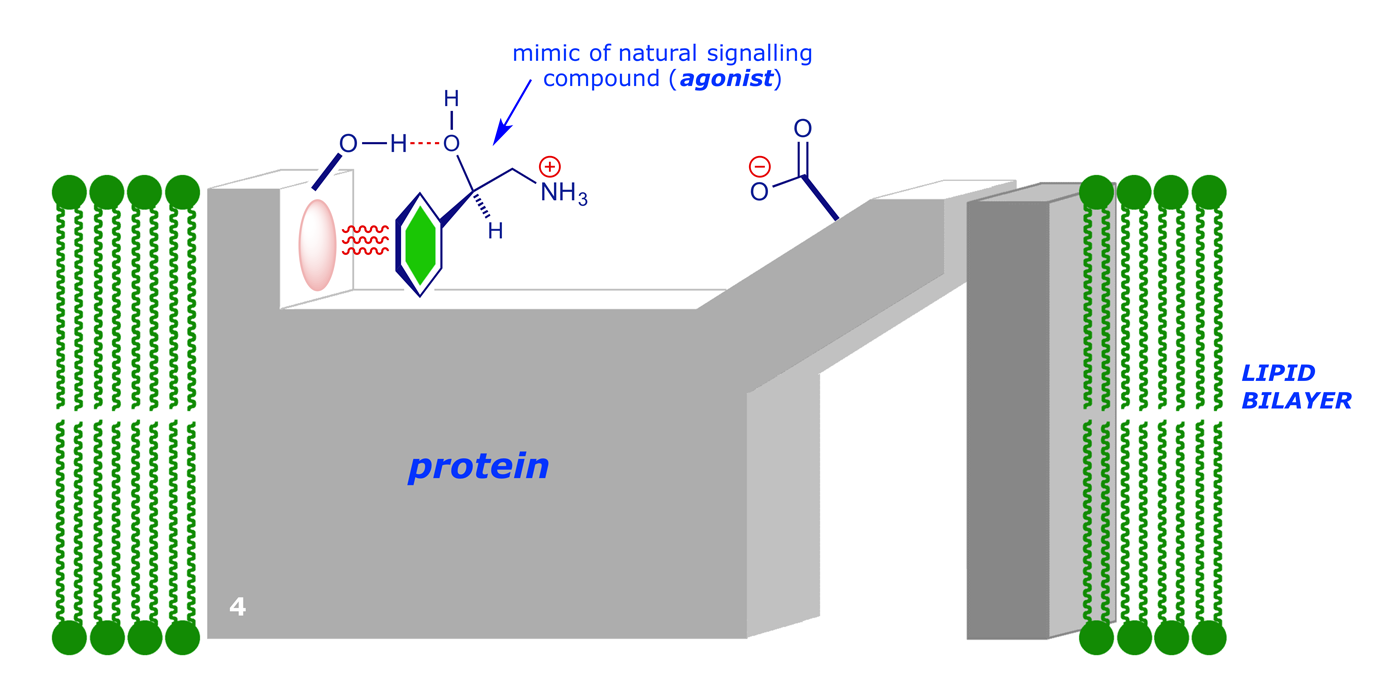
In this case, we have a modified the benzene ring (now solid green, e.g. with extra substituent groups or perhaps changed to a pyridine ring). With this new compound bound to the receptor site, the conformational change will probably still occur, and the channel will open:
So in order to produce an effective drug that will bind to the receptor (and thereby prevent the natural signalling compound from binding), but not open the channel, we need to produce a molecule that binds strongly without causing the conformational change:
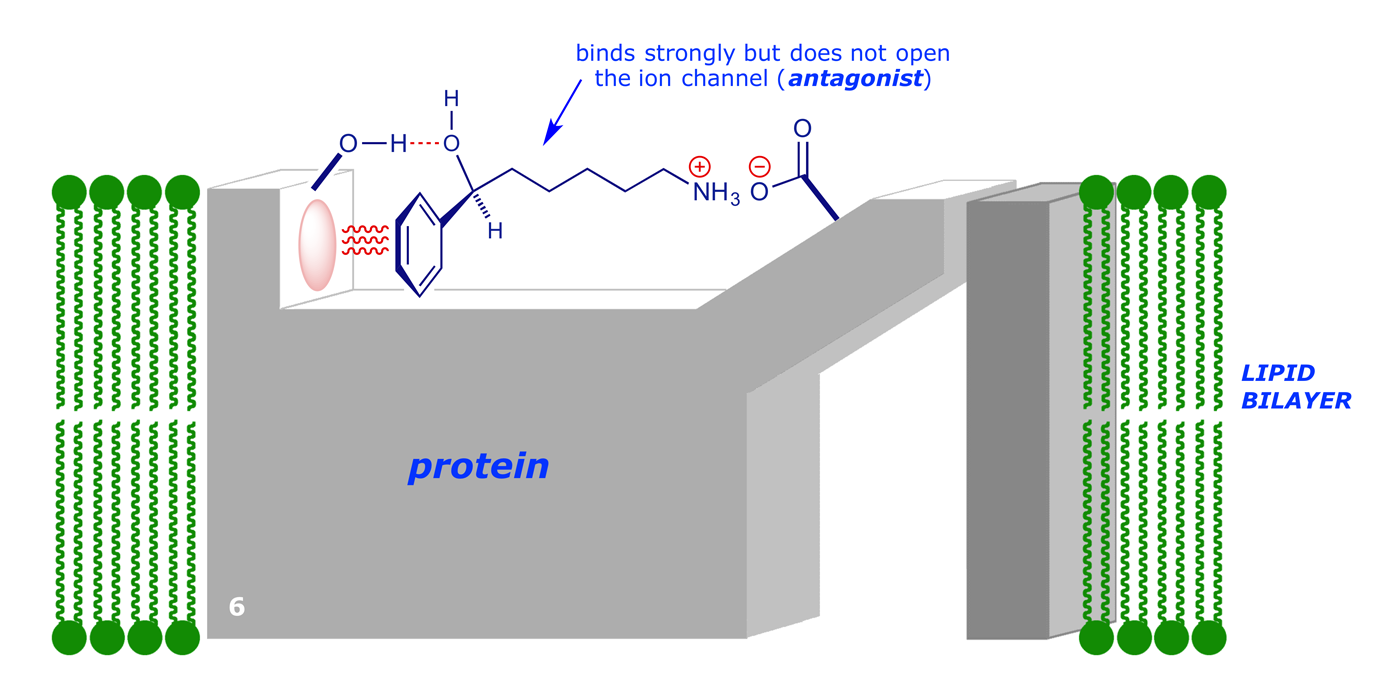
4 Cimetidine (Tagamet®) and Ranitidine (Zantac®)
Duodenal and gastric ulcers (collectively known as peptic ulcers) can affect large numbers of people who are otherwise relatively fit. Ulcers, which are usually caused by bacterial infection, are localised erosions of the mucous membrane of the duodenum or stomach, which exposes the underlying layers of gut tissue to the stomach acid. One approach to the treatment of peptic ulcers is to reduce the production of acid in the stomach. In principle this can be achieved by consuming basic materials (e.g. sodium bicarbonate), but the quantities required (ca. 60 g per day, continuously administered) mean that this is not possible without considerable discomfort. An alternative approach is to block one (or more) of the three key receptor sites on the parietal cells, and thereby stop (or reduce) hydrochloric acid production.
The parietal cells produce acid (HCl) as part of the digestion process. The production of HCl is stimulated by chemical signals which act on receptors on the surface of the parietal cells. It is now known that three signalling compounds can cause the cells to start HCl production by binding to the appropriate receptor.

Anti-cholinergic drugs (which block acetylcholine transmission) can reduce stomach acid production. However, their use is severely limited because acetylcholine is a signalling agent for a number of other bodily functions and these drugs are not usually selective, leading to a range of side-effects — dryness of mouth, urinary retention, blurred vision.
Of the other two signalling molecules, by far the most success has been achieved by blocking the action of histamine. Histamine is known to have two major roles as a signalling agent. Firstly, it can cause localised inflammation (as in the case of an allergic response) by acting at one type of receptor site (H-1 receptor), and the blocking of this site by anti-histamines is the main treatment for allergy (e.g. hay-fever). The second major function of histamine is to activate stomach acid secretion, in this case by acting at the receptor on the parietal cells (H-2 receptor). Because the H-1 and H-2 receptors are significantly different in structure (although they both have the same signalling compound), it has been possible to develop drugs which selectively act on only one or the other.
Cimetidine, an anti-ulcer drug, was the first histamine H-2 receptor antagonist and one of the first drugs to be developed on the basis of a rational design approach. The drug was developed in the UK by Smith, Kline and French (SKF; later GlaxoSmithKline) in a project led by James Black, Robin Ganellin and others.

By 1964 it had been established that histamine stimulated the secretion of acid by the stomach, but since the antihistamine compounds of the day had no effect on this process, it was reasoned that there must be another receptor that responds to histamine. The SKF group instigated a search for inhibitors of the so-called (but at that time theoretical) histamine H-2 receptor. Cimetidine was marketed 12 years later, in 1976 under the name Tagamet, and became the first 'blockbuster' drug (sales exceeding $1 billion in one year). Glaxo were also on the same search, and launched their H-2 receptor antagonist ranitidine (Zantac) in 1981. By 1988 Zantac was the world no. 1 prescription drug.
The SKF group's first lead compound was Nα-guanylhistamine. The molecule was unsuitable for drug use, but further development provided burimamide, which was 100x more active, although still not suitable for oral administration, being poorly active at physiological pH. Burimamide was modified as follows:
- Addition of a sulfide group close to the imidazole ring, to make it slightly less basic
- Addition of methyl group to the 4-position on the imidazole ring to favour the tautomeric form
of the imidazole ring that binds more strongly to the H-2 receptor
These changes gave metiamide, which has increased bioavailability and was 10x more potent than burimamide in inhibiting histamine-stimulated release of gastric acid. Clinical trials confirmed that metiamide was beneficial, but decreased white blood cell count was a frequent side-effect. The S atom was changed back to N, and an electron-withdrawing group was added to reduce the pKa of the guanidine group to the required level. Cimetidine was the eventual therapeutic agent.

5 Ion channel receptors
We have previously seen how the process of ligand-binding through induced fit can elicit changes in the tertiary structure of a receptor protein, and how, in appropriate circumstances, this might open up an ion channel, i.e. allow polar species to pass through the hydrophobic lipid bilayer and into (or out of) the cell. This is often described as a lock gate mechanism; the structures, conformational changes, and signal transmission processes in a real ion channel are complex and subtle, but the principles are well illustrated by the cartoon version shown above.
6 G-Protein-coupled receptors
Many important drugs act by binding to G-protein-coupled receptors, a group that includes the muscarinic, adrenergic and opioid receptors. The receptors mediate the activation of G proteins, which in turn control (activate, deactivate) membrane-bound enzymes. In this way G-protein-coupled receptors influence the reactions that take place inside the cell.
The receptor protein is embedded in the cell membrane, with the messenger binding site on the outer surface. On the inner surface is another binding site which is normally closed (graphic below, frame 1). When the ligand binds the receptor changes shape, opening up a binding site on the inner surface (frame 2); the G protein can then bind (frame 3).
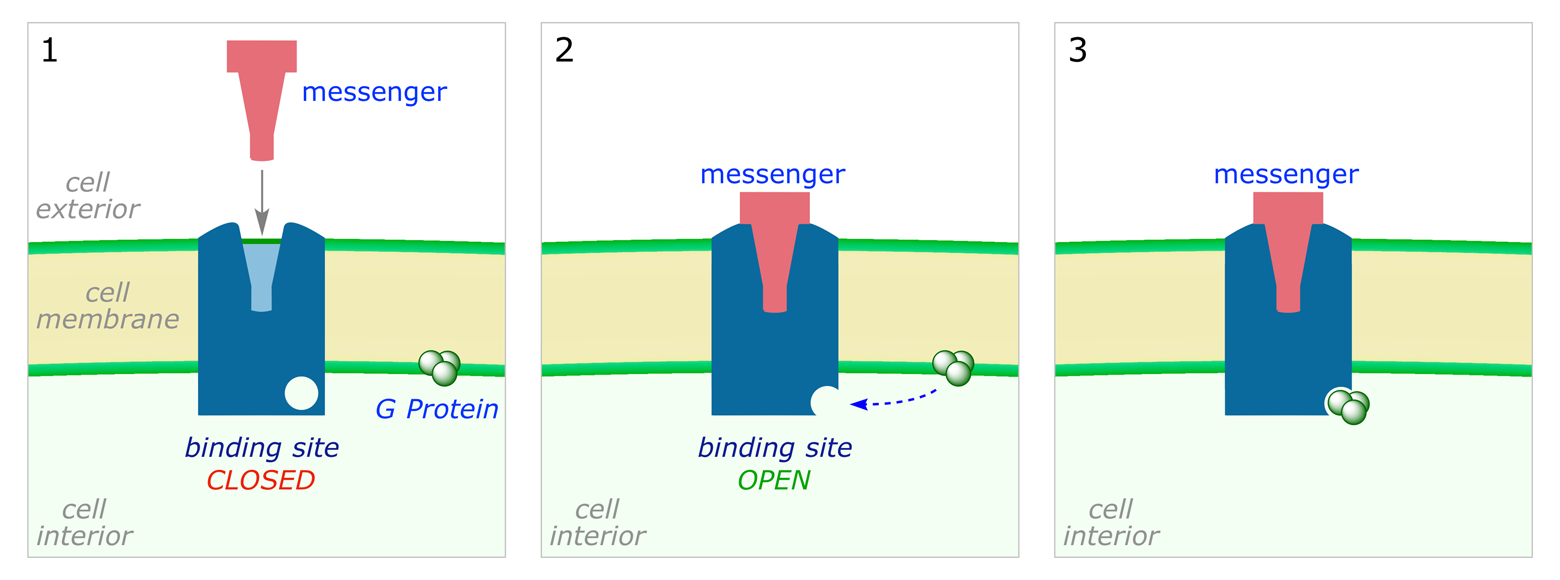
In its bound form the G protein is destabilised and fragments (frame 4). One of the sub-units travels through the membrane to a membrane-bound enzyme, where it binds to an allosteric binding site (frame 5). This causes the enzyme to change its shape, resulting in the opening of an active site and initiating a new reaction in the cell (frame 6).

Some G-protein-coupled receptors have an inhibiting effect on the bound enzyme, i.e. cause the closure, rather than the opening, of the binding site in frame 5. One activated receptor can activate several G proteins, and the activation of one enzyme by a G protein subunit (frame 5) can initiate several enzyme-catalysed reactions, so the signal is amplified in such processes.
Each G-protein-coupled receptor is folded, with seven transmembrane sections. The binding site for the G protein involves the C-terminal chain and the nearby variable intracellular loop. The exact position of the external (receptor docking) site can vary considerably.

7 Kinase-linked receptors
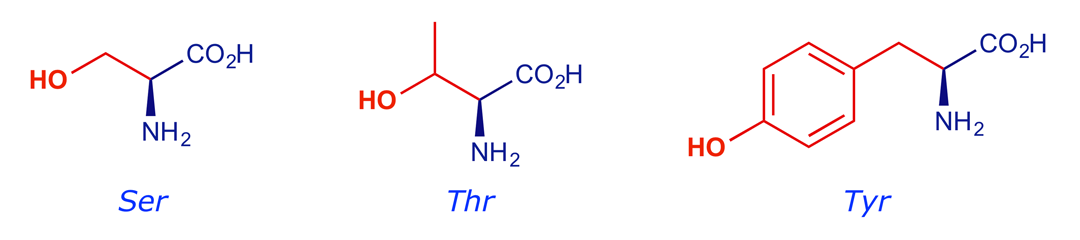
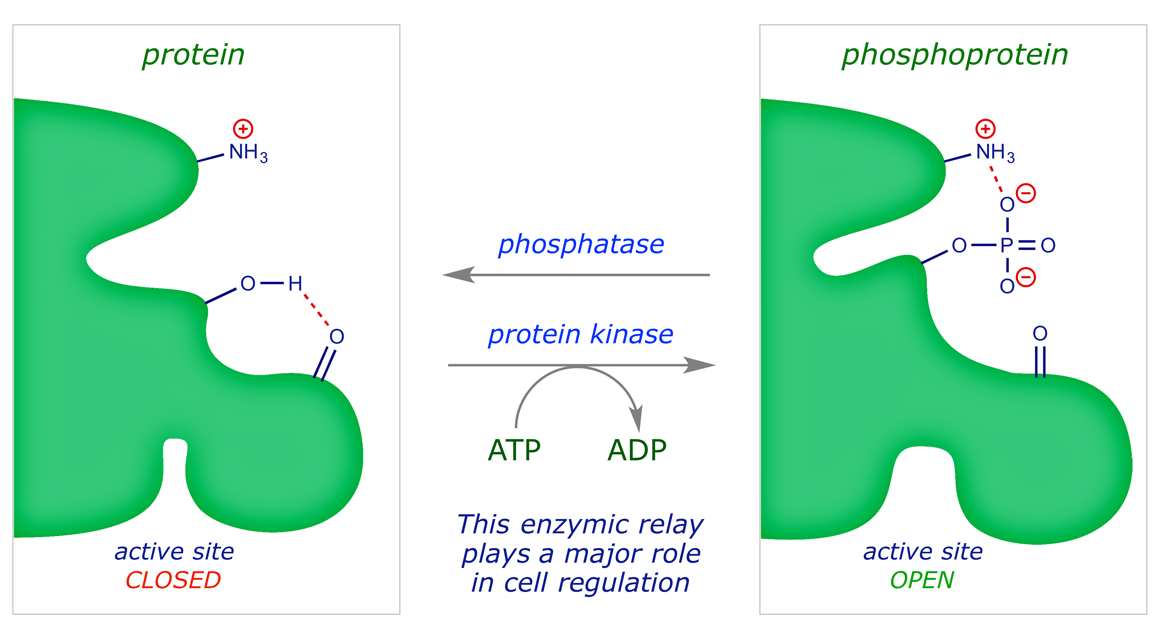
Kinase enzymes play a crucial role in the cell by phosphorylating amino acids with side-chain hydroxyl groups (serine, threonine, tyrosine). Phosphorylations of this type, which switch on (or off) an enzyme that is inside the cell, are subject to external control through receptors. Kinase-linked receptors activate enzymes directly, without the involvement of a G protein.
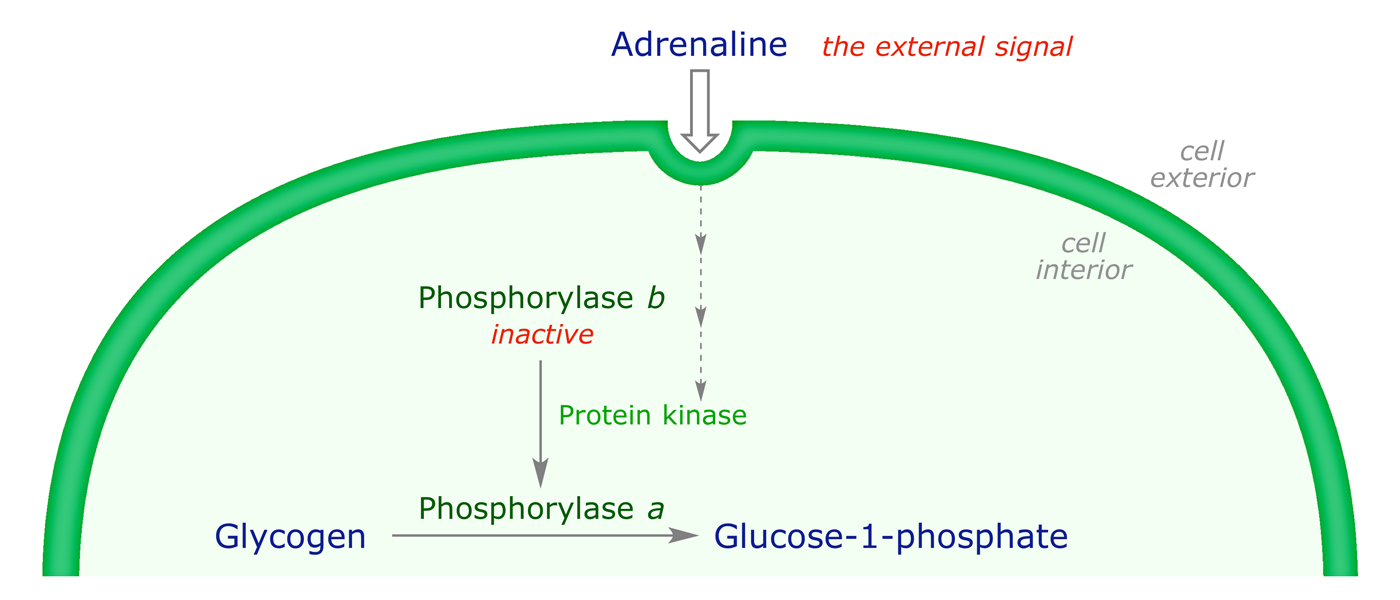
For example, the hormone adrenaline triggers a signalling sequence that results in the activation of the protein kinase enzyme that phosphorylates an inactive protein called phosphorylase b, generating its active form, phosphorylase a. The latter catalyses the breakdown of glycogen into glucose. Some enzymes are deactivated by phosphorylation.
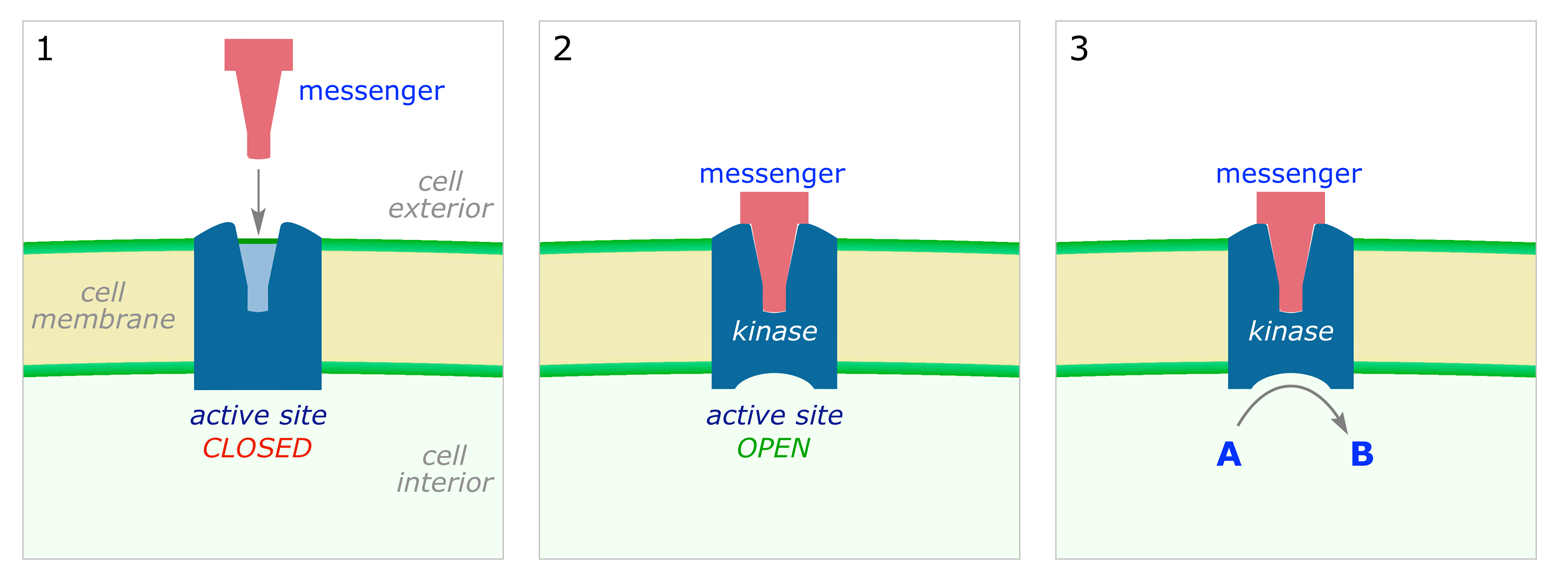
Binding of the chemical messenger to the receptor causes the protein to change shape and the active site located inside the cell is switched on. The enzyme is active while the messenger is bound; one messenger molecule can mediate many phosphorylations.
Receptor tyrosine kinases are both receptor and enzyme in one protein. The receptor protein is embedded in the cell membrane, with extracellular and intracellular components. The outer surface contains the binding site for the messenger, while the intracellular part has the catalytically active binding domain whose normal resting state is 'off.'
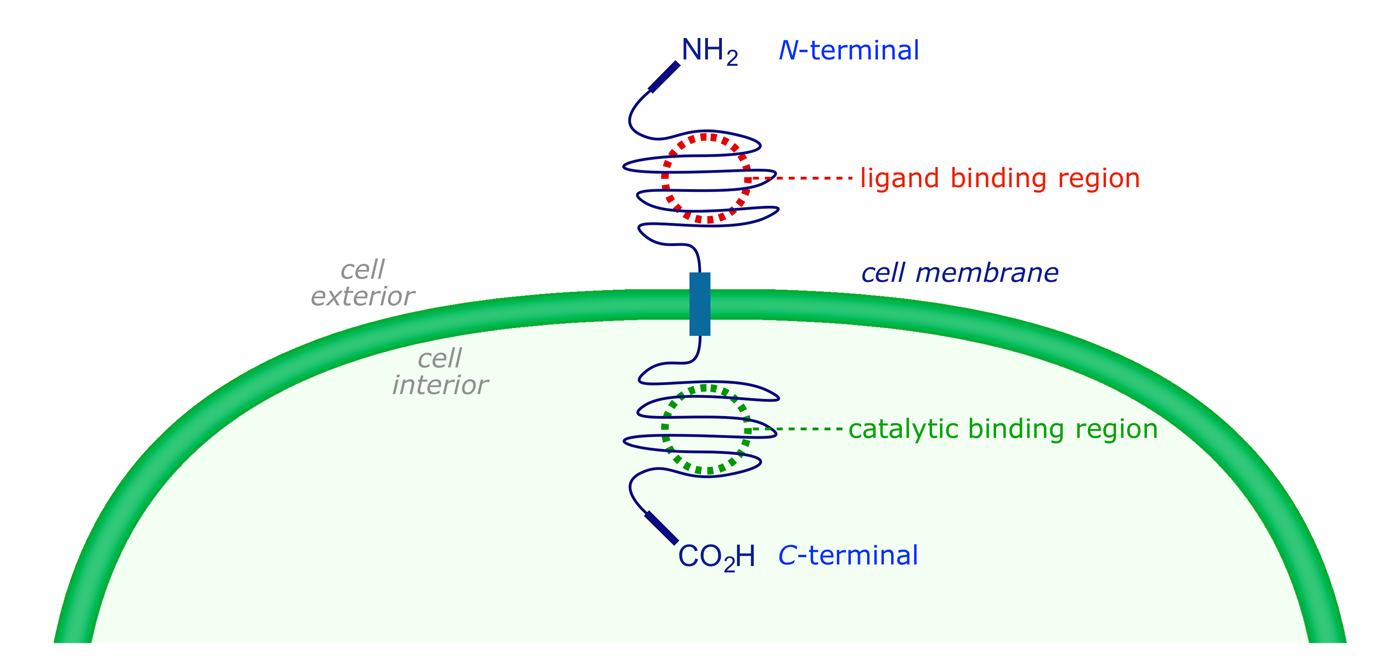
The catalysed reaction is the phosphorylation of a protein's tyrosine residue, requiring ATP as a cofactor (source of phosphate). Tyrosine kinases are activated by numerous hormones and growth factors, and play an important role in regulating cell division. Overexpression or loss of control of these kinases can result in malignant growth disorders, i.e. cancers.
8 The development of imatinib (Gleevec®)
Tyrosine kinase enzymes are key regulators of cell growth processes. They are targeted by a variety of new anticancer drugs, and the first protein kinase inhibitor to reach the market was imatinib (Gleevec®), a landmark in anticancer therapy and the first drug to target a molecular structure that is unique to a cancer cell. It acts as a selective inhibitor for a hybrid tyrosine kinase known as Bcr-Abl which is active in certain tumour cells.
8.1 Chronic myelogenous leukemia
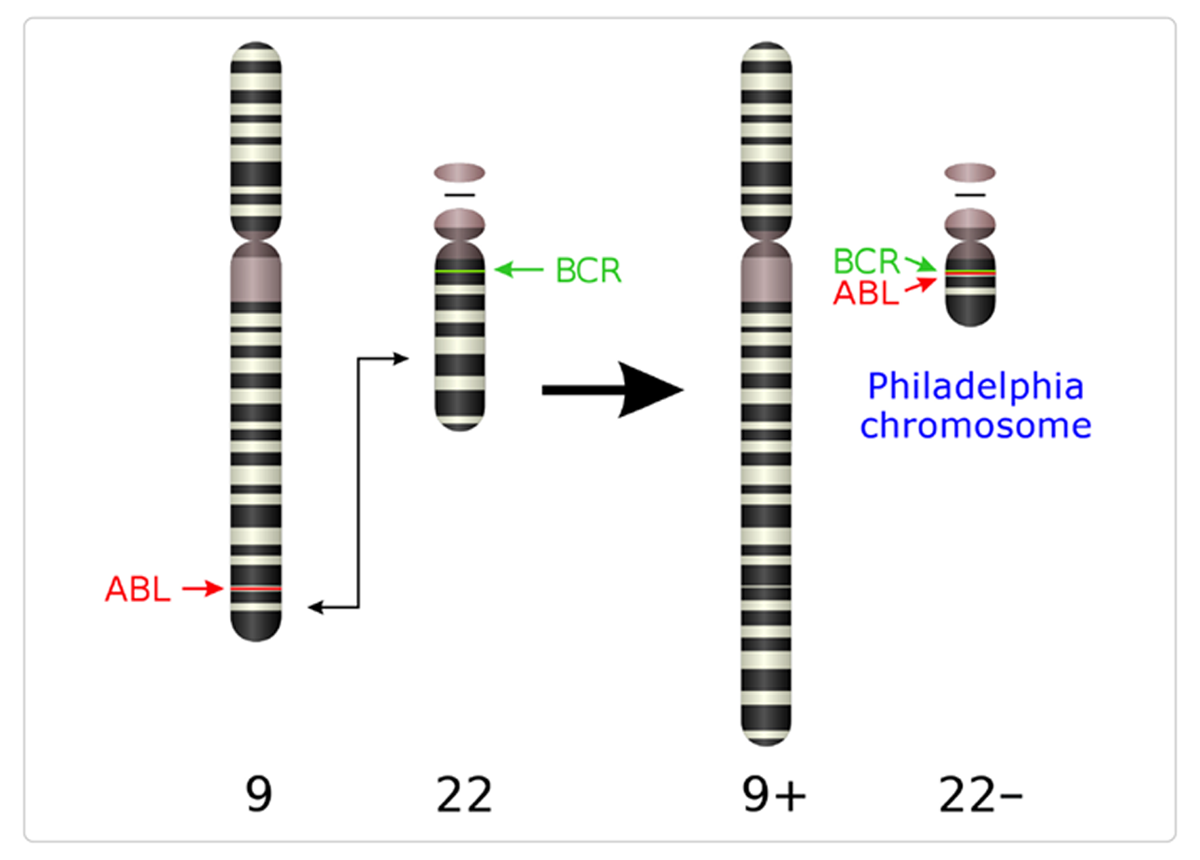
Chronic myelogenous leukemia (CML) is a characteristic of the presence of the Philadelphia chromosome (Ph), a genetic abnormality resulting from a reciprocal translocation of genes between chromosomes 9 and 22 which fuses portions of the genes encoding the proteins Bcr and Abl. The abnormal Bcr-Abl fusion gene encodes a tyrosine kinase whose 'resting state' is ON rather than OFF (Abl in normal cells has tightly controlled activity). The Bcr-Abl protein can transform cells, making them growth-factor independent and causing malignancy.
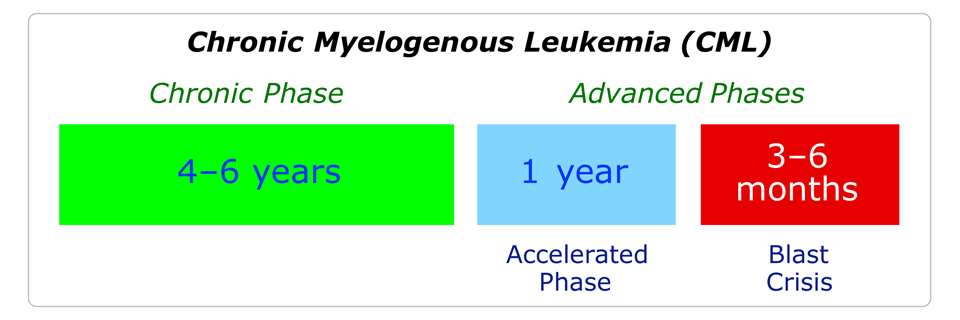
CML has three phases: chronic phase, accelerated phase and blast crisis. The median survival for the blast crisis phase is 3–6 months. Inhibitors of the Bcr-Abl tyrosine kinase were sought by Novartis using rational drug design principles, and they launched imatinib (Gleevec®) in 2001. In a clinical trial this compound led to complete responses in 96% of patients.
8.2 Optimisation of the drug structure
The lead compound for the development of imatinib was the phenylaminopyrimidine I, which was identified using high-throughput screening for tyrosine kinase inhibitors. The initial aim of the search was to find inhibitors of another protein kinase, protein kinase C (PKC), but the hit 'scaffold' proved easy to adapt. Simple modifications revealed that selectivity for tyrosine kinases was possible; imatinib emerged from the development sequence shown below.

Key structures in the lead optimisation process:
II Adding a 3'-pyridyl (blue) at the 3'-position of the pyrimidine enhanced cellular activity.
III An amide group (red) attached to the phenyl ring gave activity against tyrosine kinases.
V The 'flag-methyl' group (gold) attached to the diaminophenyl ring abolished unwanted activity against PKC by inhibiting rotation about the C–N bond (shown in bold).
VI The final attachment of an N-methyl piperazine moiety (green) significantly increased both the solubility and the oral bioavailability.
The ortho methyl group in V serves as a conformational blocker — this compound has enhanced activity against tyrosine kinases but none against serine-threonine kinases, which showed that the methyl group favoured a conformation suitable for binding to tyrosine kinases but not to serine-threonine kinases. The pyridine and pyrimidine rings locate as far away as possible from the methyl group for steric reasons. Further modifications were then carried out to maximise activity and selectivity, including the addition of a piperazine ring. This is also important for water solubility, as it contains a basic N which allows the formation of water-soluble salts. A one-carbon spacer was placed between the aromatic and piperazine rings.
The X-ray crystal structure of imatinib inside the target protein shows that the amide acts as an anchoring group. The amide forms H-bonds to the residues Glu-286 and Asp-381, both of which are essential to the catalytic mechanism of the enzyme. Another H-bond links an amino group in imatinib and the 'gatekeeper' Thr-315 residue in the active site (there is no activity if this amino group is alkylated). Selectivity is also favoured by the ortho methyl group that was introduced as a conformational blocker. The methyl group is able to bind to a hydrophobic pocket that would be inaccessible if a larger gatekeeper residue were present.

8.3 General synthesis of imatinib and analogues
The synthesis of imatinib and its analogues involves the pyridine structure A as starting material. The ketone A is first transformed to B, and the latter is condensed with the phenylguanidine derivative C. This generates the central pyrimidine ring in D. The remaining two steps involve reduction of the aromatic nitro group to the corresponding amine E, and then acylation to give the final amide product F. This route allows the synthesis of a large variety of amides from intermediate E.

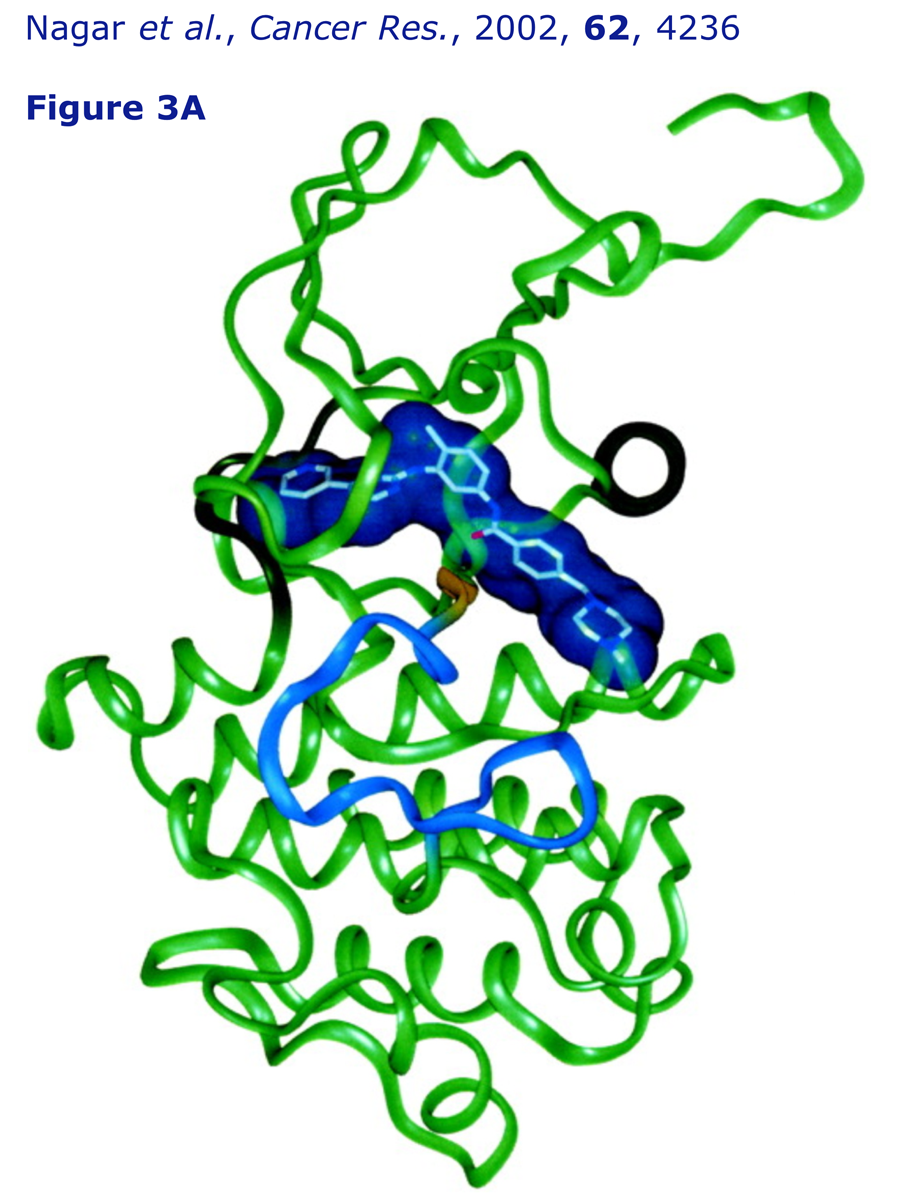
8.4 X-Ray analysis of the Gleevec binding site
An interactive 3-D molecular model of imatinib (Gleevec) complexed to Abelson tyrosine kinase, its target, is provided. The structure is based on the X-ray data published by Kuriyan and coworkers in 2002.
Crystal structures of the kinase domain of c-Abl in complex with the small molecule inhibitors PD173955 and Imatinib (STI-571)
B. Nagar, W. G. Bornmann, P. Pellicena, T. Schindler, D. R. Veach, W. T. Miller, B. Clarkson and J. Kuriyan, Cancer Res., 2002, 62, 4236–4243.
Critical to the binding of imatinib is the adoption by the kinase of an inactive conformation, in which the centrally located 'activation loop' is folded up inside the protein structure, with the enzyme's key tyrosine (Tyr-393) not phosphorylated. The drug occupies the normal location of the cofactor, ATP, so not only is the structure inactive, but phosphorylation (i.e. activation) cannot take place.
Imatinib (Gleevec)