
The ACE Inhibitors
- The development of the ACE inhibitors
- Mechanism of action
- X-Ray analysis of the captopril binding site
- ACE inhibitors after captopril
1 The development of the ACE inhibitors
Angiotensin converting enzyme (ACE) is part of the renin-angiotensin system and its main role in the body is to break down angiotensin I (decapeptide) into angiotensin II (octapeptide). It does this by cleaving the second amide bond in from the carboxyl terminus.
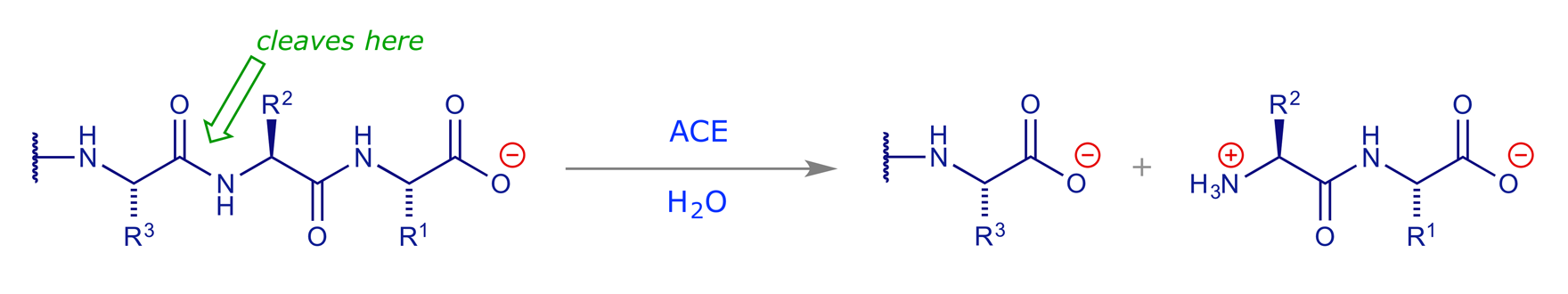
The product (angiotensin II) then triggers a series of events which lead to an increase in blood pressure. In patients that suffer from abnormally high blood pressure (hypertension), stopping the formation of angiotensin II by blocking the action of ACE can be an effective remedy.

The 3-D structure of ACE has been determined by X-ray crystallography, but the first inhibitors of this enzyme were developed while it was still unknown. The area was pioneered by Cushman, Ondetti and Rubin at Squibb (now Bristol-Myers Squibb) in the mid-1970s and led to the identification of several antihypertensive agents, including captopril (marketed as Capoten), Squibb's first billion-dollar drug, and one of the earliest successes of structure-based drug design. The story of captopril is one of the most illuminating case studies in the history of drug development, and the following discussion draws strongly on two of the associated research papers:
1. Design of potent competitive inhibitors of angiotensin-converting enzyme. Carboxyalkanoyl and mercaptoalkanoyl amino acids [D. W. Cushman, H. S. Cheung, E. F. Sabo and M. A. Ondetti, Biochemistry, 1977, 16, 5484–5491].
2. High-resolution crystal structures of Drosophila melanogaster angiotensin-converting enzyme in complex with novel inhibitors and antihypertensive drugs [M. Akif, D. Georgiadis, A. Mahajan, V. Dive, E. D. Sturrock, R. E. Isaac and K. R. Acharya, J. Mol. Biol., 2010, 400, 502–517].
The Squibb group started out with a clue from a natural product. The toxic effects of venom from a Brazilian viper (Bothrops jararaca) were found to be due to a sudden, massive drop in blood pressure. It was established that the compound responsible, a nonapeptide that was later named SQ20881, was a potent ACE inhibitor.

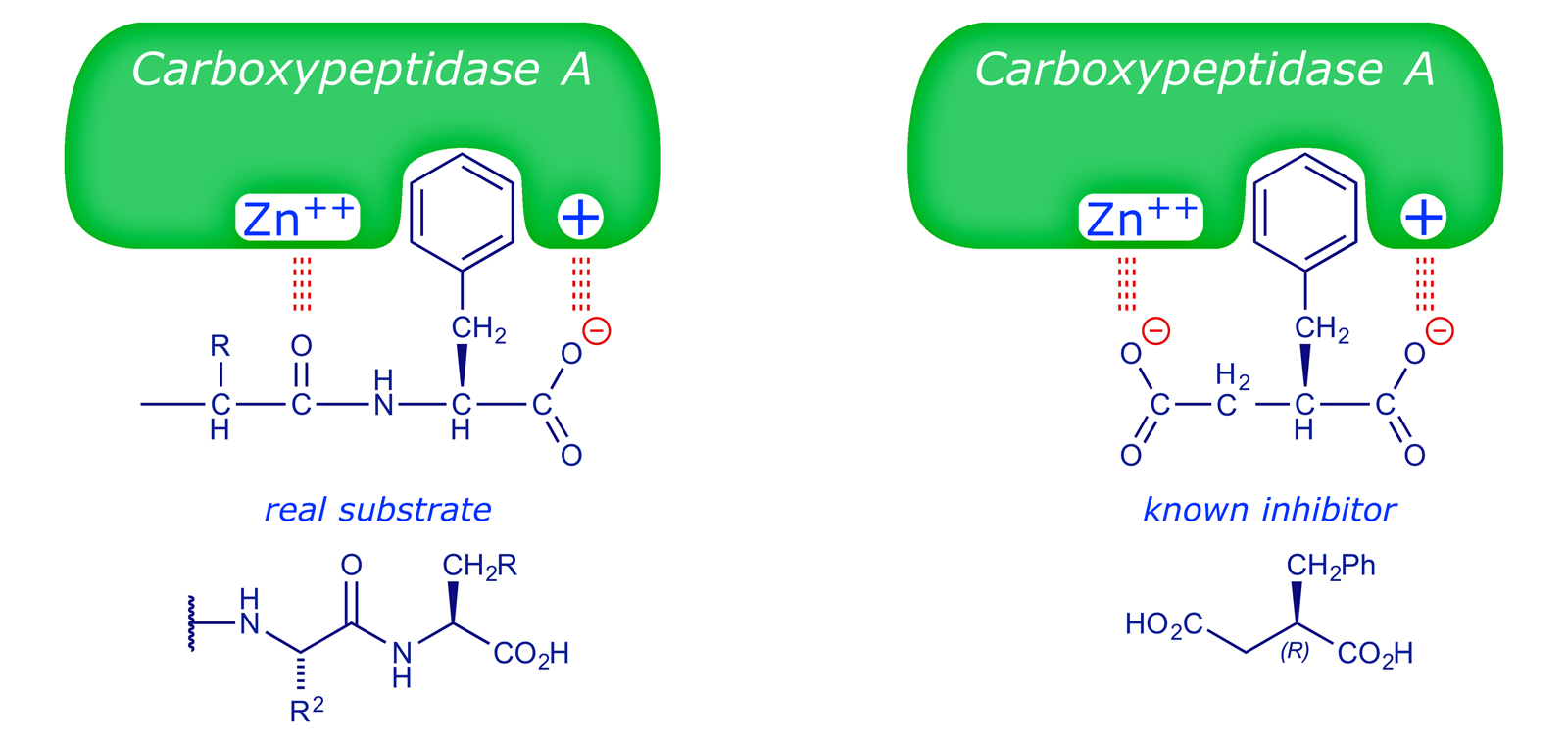
In rationalising their search for drugs with the same property, the Squibb team advanced a hypothetical model of the active site of ACE based on comparisons with another zinc metalloproteinase, carboxypeptidase A. This enzyme, present in the human digestive system, is able to cleave the C-terminal amino acid from a peptide chain:
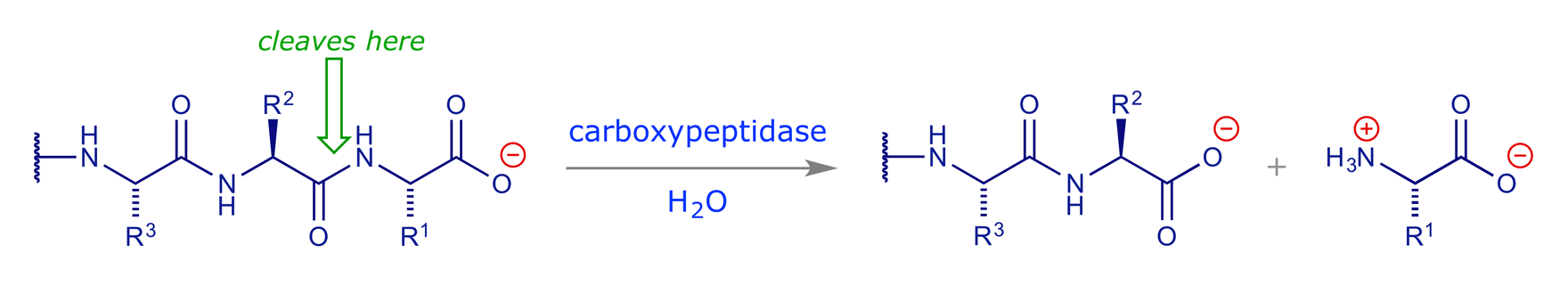
The structure of carboxypeptidase A was established in the 1960s and it was known that (R)-2-benzylsuccinic acid functioned as an inhibitor of this enzyme. The Squibb rationale is summarised in the following figures (adapted from ref. 1).
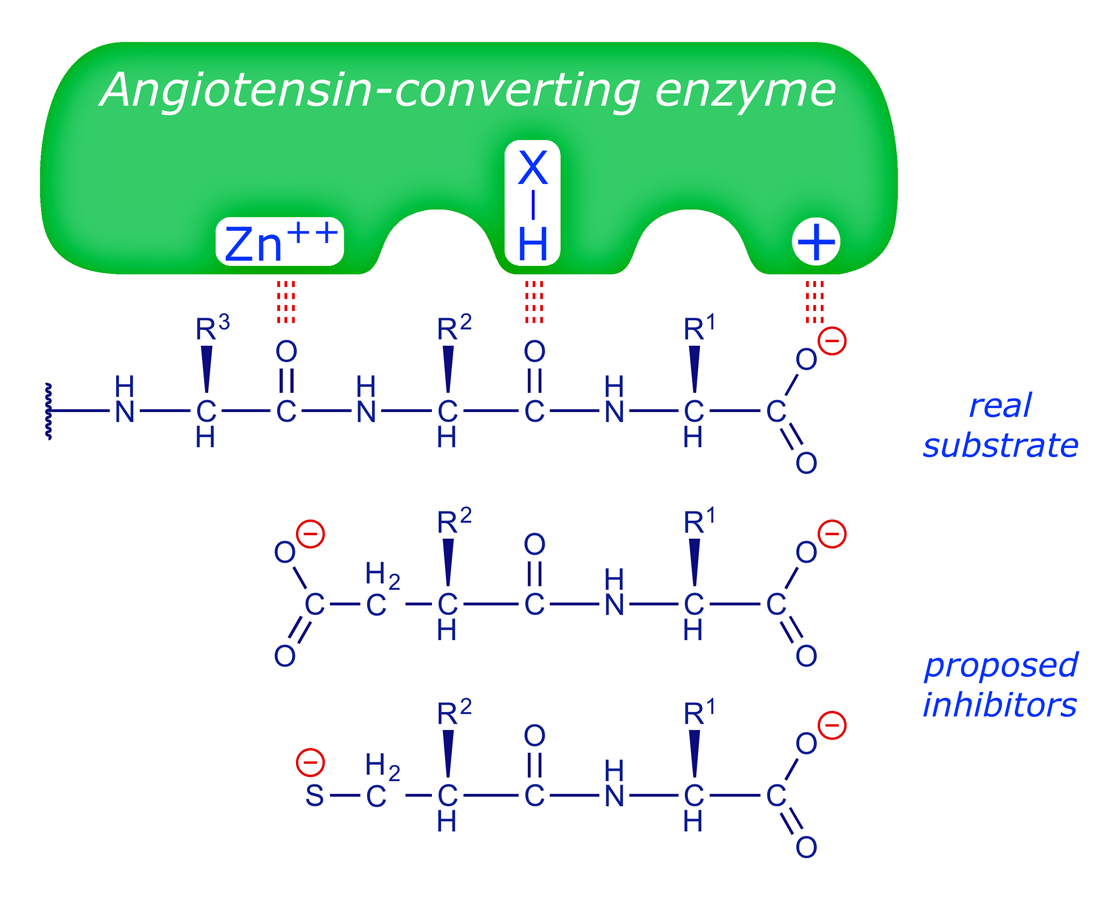

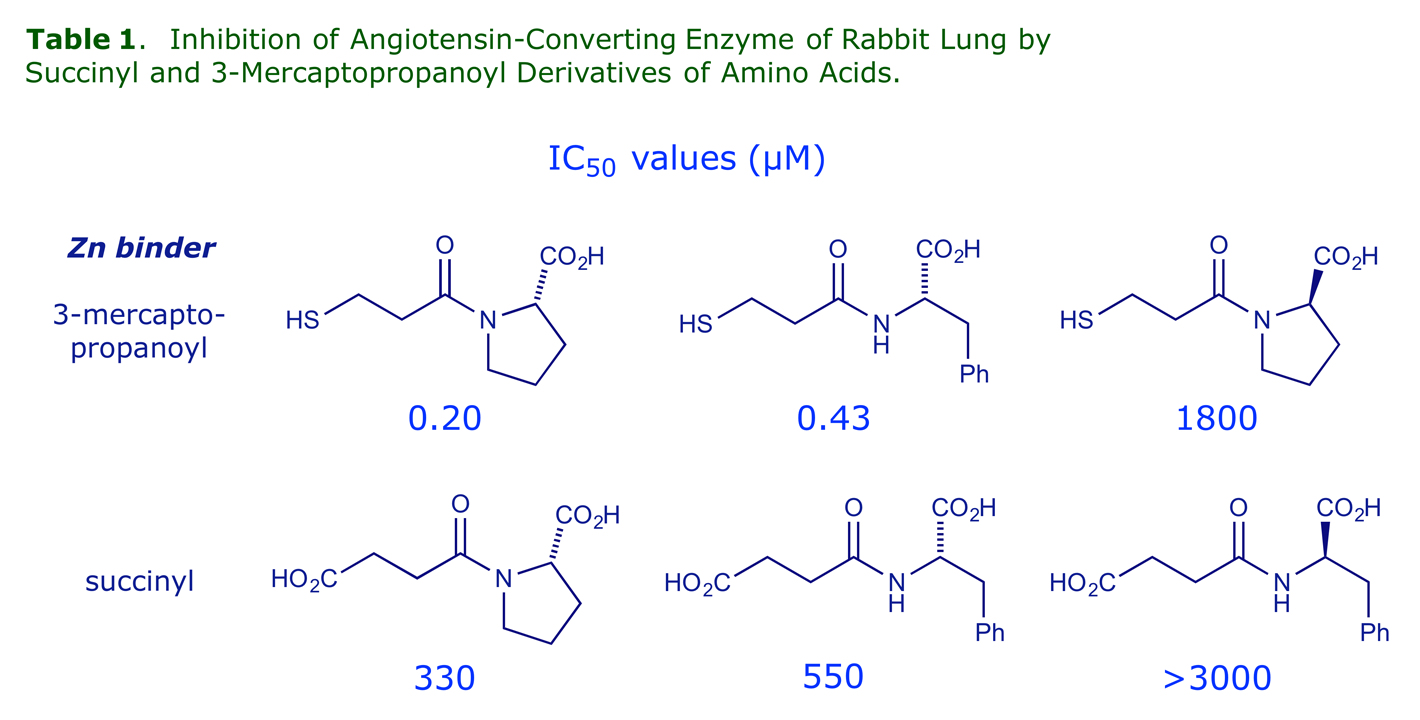
The chemists at Squibb made a series of amino acid derivatives which were tested as ACE inhibitors. The following tables show some of the structure-activity relationships (SARs) they found, and reveal the logic of the drug design process, whereby a structure is systematically modified until it possesses the optimum properties (N.B. only a few of the many structures tested are shown here).
The best terminal unit is proline (cf. the first 'hit' compound SQ20881) and it must have the (S)-configuration. A 3-mercaptopropanoyl side-chain is a more effective Zn binder than carboxyl.
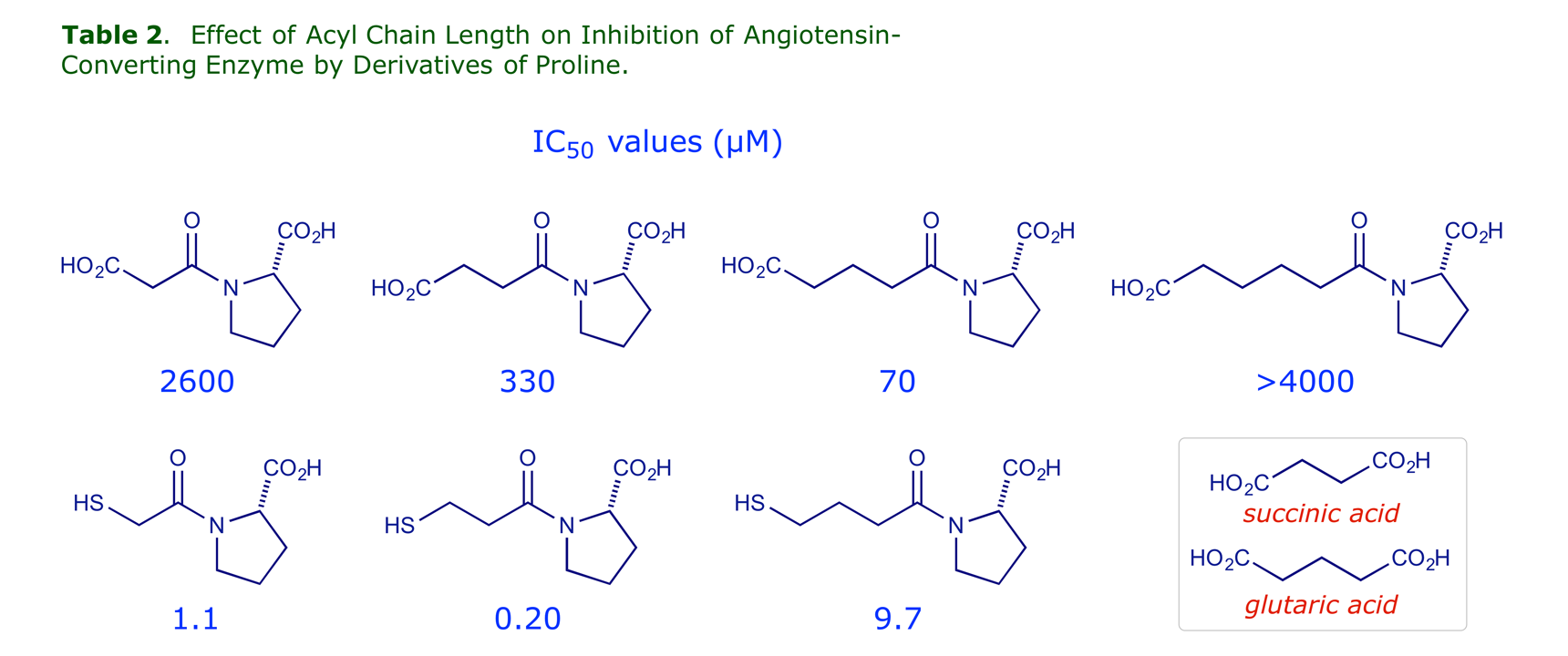
The most effective spacer for a mercaptoacyl Zn binder is –(CH2)2– whereas for a carboxyl binder the –(CH2)3– is better (glutaroyl is more effective than succinyl):
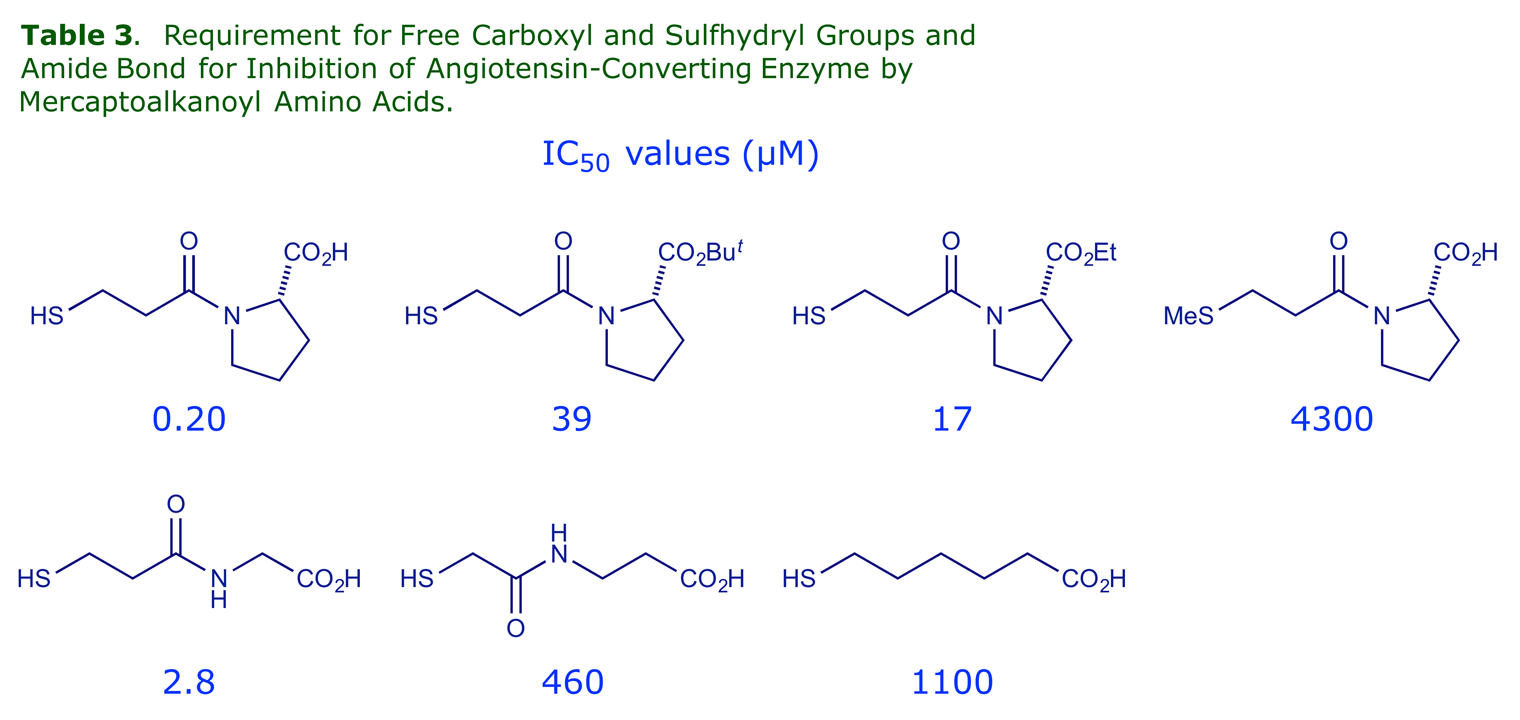
For optimum activity, the mercaptoacyl Zn binder should have a free –SH group, the proline should have a free carboxyl, and a central amide link with the correct orientation is needed.
The inhibitor constants of the most potent of the compounds tested, including the snake venom peptide SQ20881, are shown in Table 4. Using Lineweaver-Burk plots it was confirmed that all were competitive inhibitors of ACE.

The competitive nature of the inhibition with respect to substrate is fully consistent with the binding of these inhibitors to the active site of the angiotensin converting enzyme.
REMINDER: If a reversible inhibitor can bind to the enzyme active site in place of the substrate, it is described as a competitive inhibitor. In pure competitive inhibition, the inhibitor is assumed to bind to the free enzyme but not to the enzyme-substrate (ES) complex. The binding is described thus:
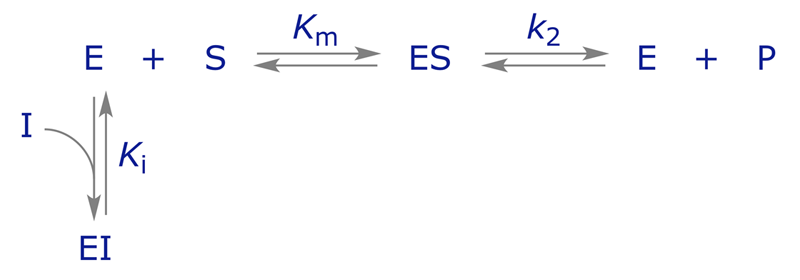
Here Ki is the dissociation constant for the enzyme-inhibitor (EI) complex. EI does not react to form E + P, and the enzyme is unable to bind both S and I at the same time.
2 Mechanism of action
The mechanism by which ACE is now believed to function is shown below. In the complete scheme, the natural substrate (angiotensin I) is cleaved at the second peptide bond in from the C-terminus to give two smaller peptides. The process is facilitated by a nearby basic group that effectively 'holds' a proton during the hydrolytic step.
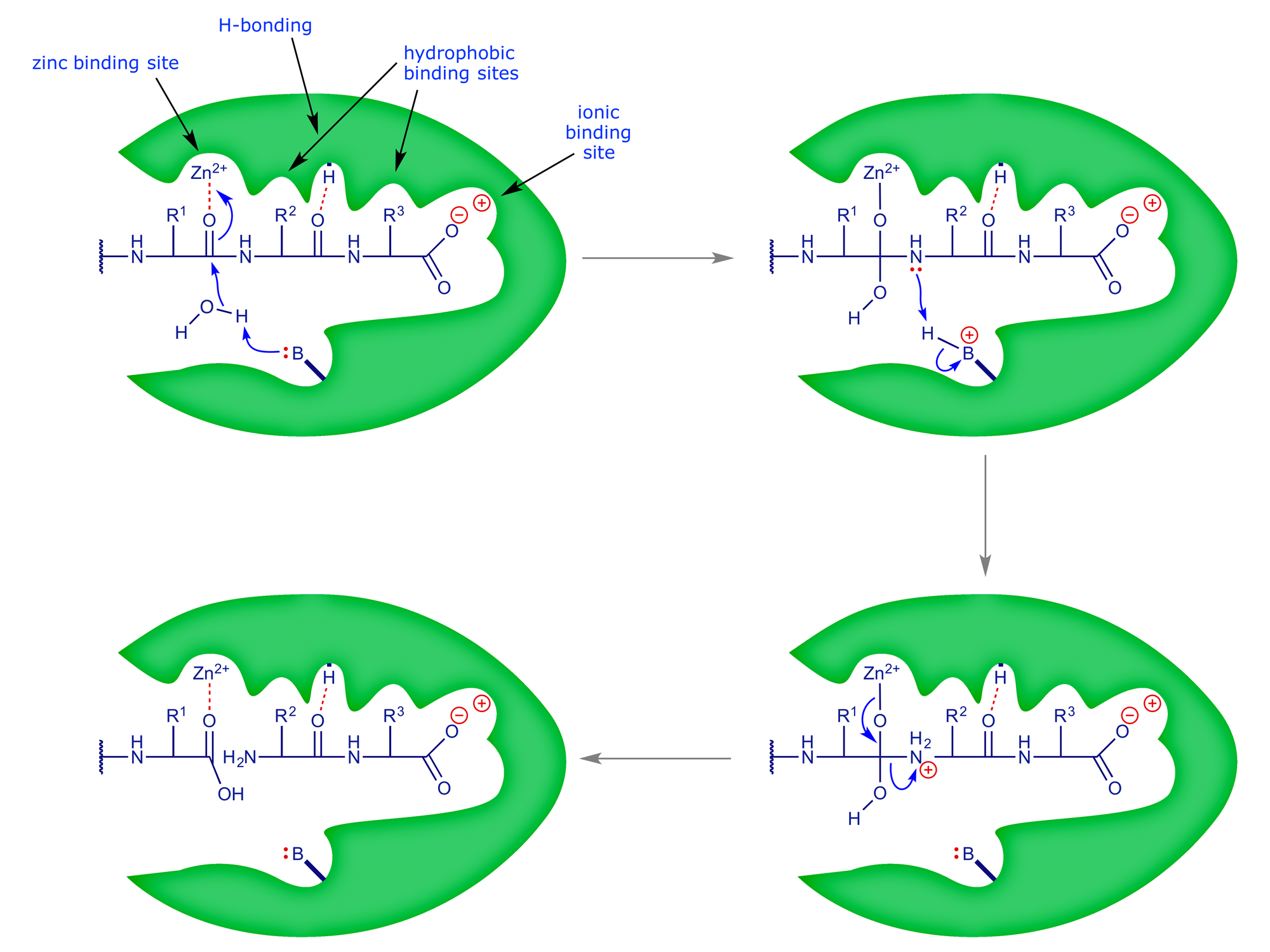
Captopril binds strongly to the active site and thereby prevents the processing of the natural substrate, angiotensin I (see Section 3 for the X-ray analysis of the binding site).

The drug structure maintains all the key binding interactions, but does not participate in the reaction. Because it binds so strongly to the enzyme active site, none of the natural substrate can bind, and so no angiotensin II can be produced — this results in a lowering of blood pressure. Captopril was marketed in 1981, and since that time a number of other ACE-inhibitors have been developed:
The adverse effect and pharmacokinetic limitations of captopril stimulated the development of enalapril and subsequent ACE inhibitors (discussed later). The Bristol-Myers Squibb patent for captopril expired in 1996.
3 X-Ray analysis of the captopril binding site
Interactive 3-D molecular models of the ACE-captopril complex are provided, based on the X-ray analyses of enzyme-drug complexes published by Acharya and coworkers in 2004 and 2010.
The binding of the antihypertensive drug captopril to human testicular angiotensin I-converting enzyme
R. Natesh, S. L. U. Schwager, H. R. Evans, E. D. Sturrock and K. R. Acharya, Biochemistry, 2004, 43, 8718–8724).
High-resolution crystal structures of Drosophila melanogaster angiotensin-converting enzyme in complex with novel inhibitors and antihypertensive drugs
M. Akif, D. Georgiadis, A. Mahajan, V. Dive, E. D. Sturrock, R. E. Isaac and K. R. Acharya, J. Mol. Biol., 2010, 400, 502–517).
Captopril
Captopril is the smallest orally active tight binding peptide analogue inhibitor of ACE. In the ACE–captopril structure the thiol group of the inhibitor makes a direct interaction with the catalytic Zn2+ ion (distance, 2.15 Å). The captopril molecule is also held by 8 H-bonds, including three with water molecules. The central carbonyl oxygen positioned between the thiol and the terminal proline moiety of captopril is anchored by two strong H-bonds with His-337 and His-497. One of the oxygen atoms from the proline moiety's carboxylate group is held by interactions with side-chains of residues Gln-265, Lys-495 and Tyr-504. The second oxygen of the proline moiety's carboxylate group in the present is held indirectly by water-molecule-mediated interactions with Asn-261 and Glu-150. Furthermore, a network of water molecules makes indirect interactions between the carboxylate oxygen and with Arg-356, Asp-360 and Gln-361 in the S1' and S2' subsites.

4 ACE inhibitors after captopril
If we relate the captopril story, thus far, to our 'drug development pipeline' (see graphic in Section 1.2), we can see the context of the cycles of synthesis and screening. The first hit was a snake venom polypeptide, which was not drug-like for PK reasons, i.e. lack of oral bioavailability (it could not reach the target via the digestive system, requiring intravenous injection). But it led the Squibb team to the lead structures A and B, and optimisation of the latter eventually provided captopril.

To optimise the ACE inhibitor profile revealed by captopril, and in particular to deal with the metallic taste problem, medicinal chemists explored the lead structure A first pursued by the Squibb group, namely the use of carboxylate, instead of sulfhydryl, as the Zn-binding group. The first of the dicarboxylate ACE inhibitors was enalaprilat 2, in which the thiol group of captopril was replaced by a carboxylate. However, enalaprilat had a problem of its own — the structural modifications led to polarity chracteristics that prevented oral administration (in tablets). Thus, it was only suitable for intravenous administration. This was overcome by the researchers at Merck by the esterification of enalaprilat to produce enalapril 3.
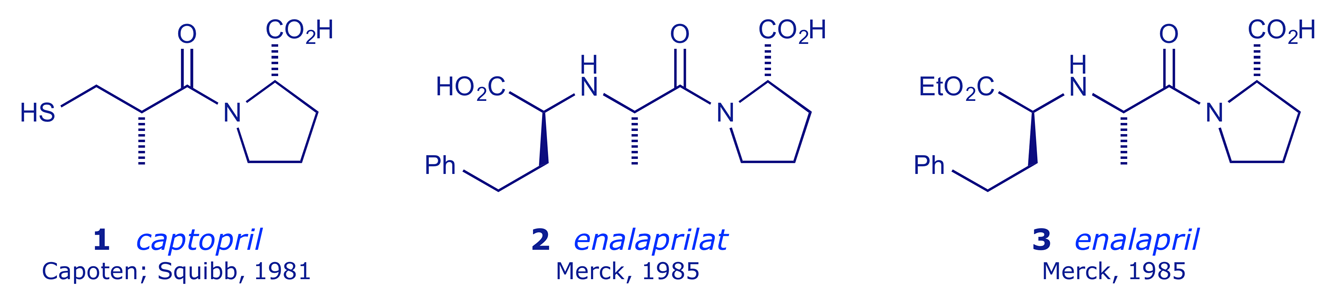
Enalapril is a prodrug — it is de-esterified in vivo to the active form (enalaprilat) by various esterases. The peak plasma enalaprilat concentrations occur 2–4 h after oral administration. Enalaprilat has effects similar to those of captopril. Enalaprilat is still available, but only for intravenous use in hypertensive emergencies. Further R & D in this area led Merck to market lisinopril 4, a more hydrophilic ACE inhibitor with a long half-life and the ability to penetrate tissue. Lisinopril is not metabolised by the liver, but is excreted intact in the urine.
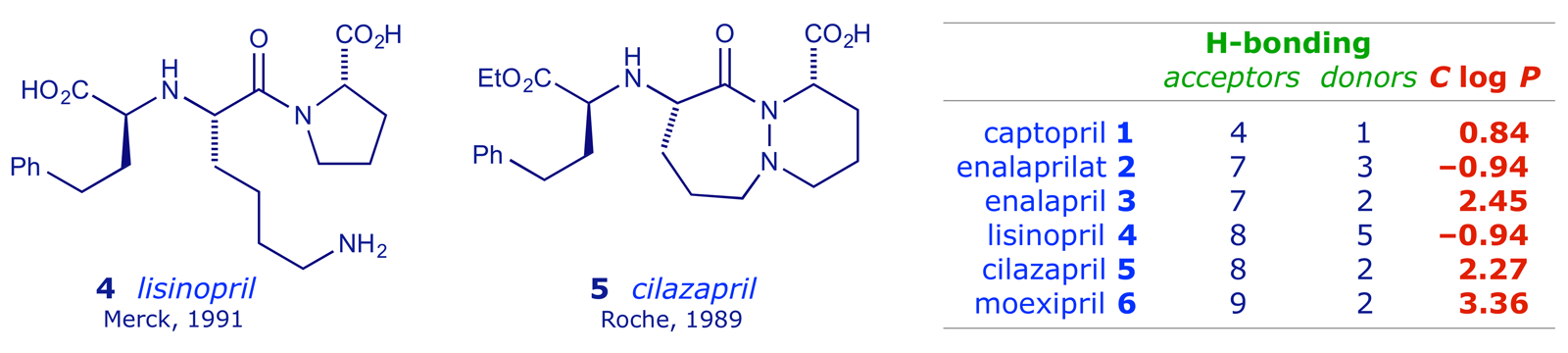
By the early 1990s, Roche had also developed cilazapril 5, which lacked the proline unit that featured in every ACE inhibitor so far. Other pro-drug structures emerged, e.g. moexipril 6, which de-esterifies in the liver to form the pharmacologically active compound (moexiprilat). Moexipril is incompletely absorbed after oral administration and its bioavailability is low. The long pharmacokinetic half-life makes it persistent, offering the advantage of once-a-day administration as a cardioprotective agent. Moexipril 6 is lipophilic (see Table) and able to penetrate membranes more readily than the hydrophilic ACE inhibitors. This means that it can counteract the effects of tissue ACE (inhibitors in the bloodstream only target plasma ACE). There is a significant reduction in tissue ACE (lung, myocardium, aorta, and kidney) activity after moexipril.
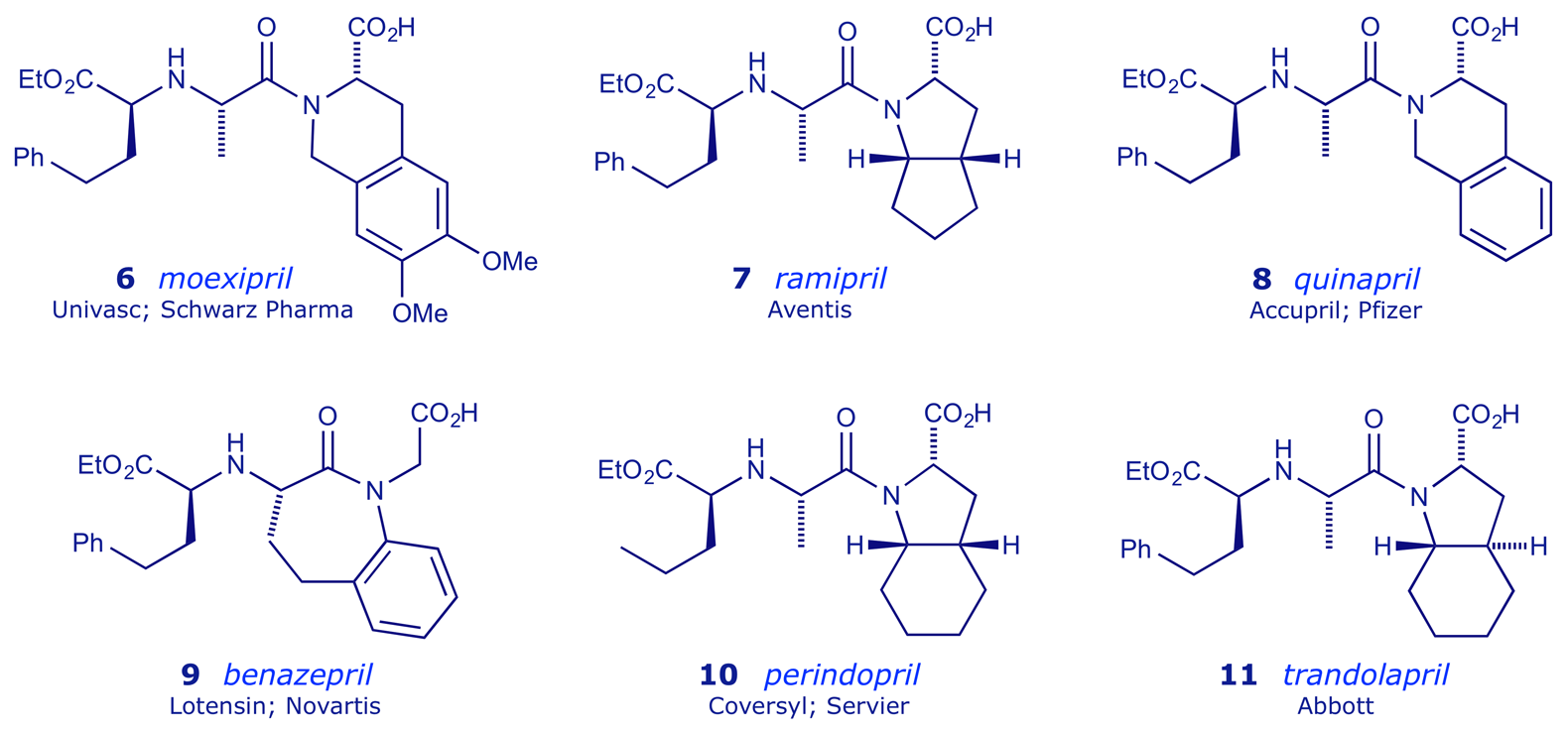
Other hydrophobic ACE inhibitor prodrugs include ramipril 7, quinapril 8 and benazepril 9. Of these, note that benazepril 9 (Lotensin®) has no structural constraint at all on the terminal carboxyl group (Squibb tried this: see Section 1 Table 3 above). Perindopril 10 has a methyl group in place of the phenyl group present in all of the other listed dicarboxylate structures.
The scale of the impact of ACE inhibitors is reflected in the following statistic: They were ranked fifth in the list of most prescribed class of drug in 2010, featuring in 168 million prescriptions.