
Lessons from Captopril
1 Drug design principles
If we relate the captopril story, thus far, to our 'drug development pipeline' scheme, we can see the context of the iterative cycles of synthesis and screening. The original hit was a snake venom polypeptide, but this was not drug-like for PK reasons, i.e. its lack of oral bioavailability (it could not reach the target via the digestive system, necessitating intravenous injection). But it led the Squibb team to two lead structures, and optimising one of these gave captopril.
A lead compound is the starting point for drug design, which has several objectives. The eventual drug should have the features listed below. We will review here the simple molecular properties that should form part of any analysis and might be exploited for the purpose of changing an active structure into a viable drug candidate.
- Desired level of biological activity
- Selectivity for the target
- Minimal or no side-effects
- Easy to synthesise at scale
- Stable
- Acceptable pharmacokinetic properties
- Non-toxic
1.1 The role of the common functional groups
The tendency, or otherwise, of functional groups to engage in bonding with biological targets (proteins or DNA) can be analysed at a simple level, as illustrated by the following examples.
Alcohol and phenol hydroxyls are common in drugs and are often involved in hydrogen bonding, the oxygen as a H-bond acceptor (HBA) and the hydrogen as hydrogen-bond donor (HBD). Bond directions must be considered; slight deviations are possible. Such bonds are generally important in drug-target binding. Converting the hydroxyl into an ether or ester would test this, as H-bonding would be disrupted in either of these derivatives.
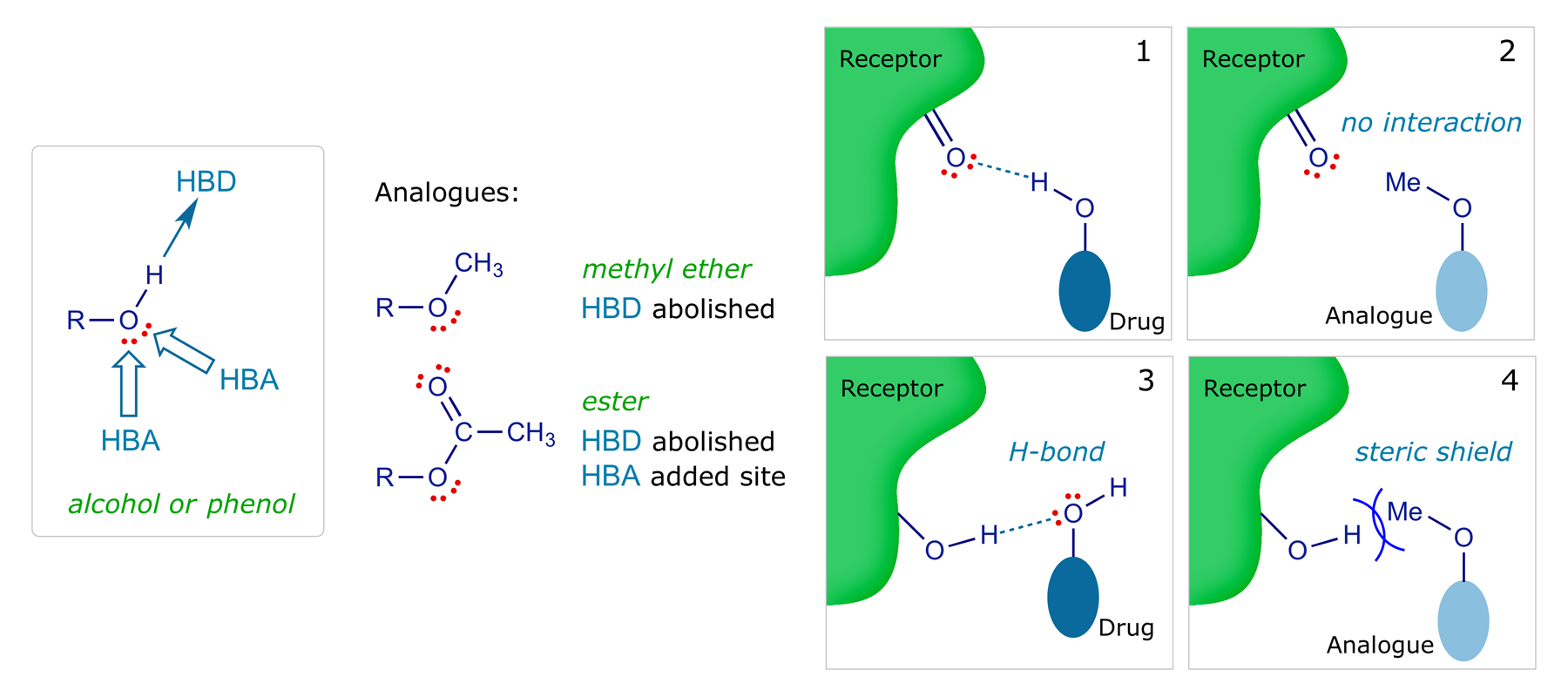
Making the ether would nullify HBD ability, and could sterically hinder or prevent the HBA process. The oxygen is still present in the ether analogue, but the extra bulk of the methyl group could hinder the close approach and be disruptive. An ester analogue would be devoid of HBD capacity, but could still act as an HBA. But the acyl group is larger than the methyl group of the ether, so this too could sterically hinder the HBA process. The electronic difference — the carbonyl is an acceptor (see resonance structure) — means that the lone pair will be less effective as an HBA. Of course, the carbonyl oxygen may act as a new HBA.
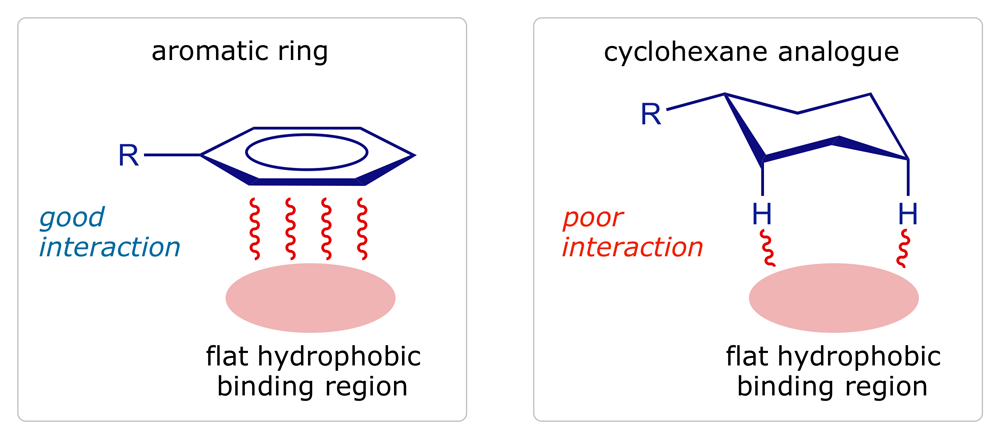
Aromatic rings are planar, hydrophobic structures, commonly involved in van der Waals and hydrophobic interactions with flat hydrophobic regions of the binding site. The saturated analogue (e.g. cyclohexane instead of benzene) may not bind so well as the ring is no longer flat — the axial protons can interact weakly, but will act as buffers, keeping the rest of the cyclohexane ring at a distance. The binding region for the aromatic ring may also be a narrow slot rather than a planar surface, in which case the cyclohexane ring would be unlikely to bind as it is too bulky. Alkenes are also planar and hydrophobic so they too can interact with hydrophobic regions of the binding site through van der Waals and hydrophobic interactions. They are often reduced to saturated systems in order to investigate their binding role.
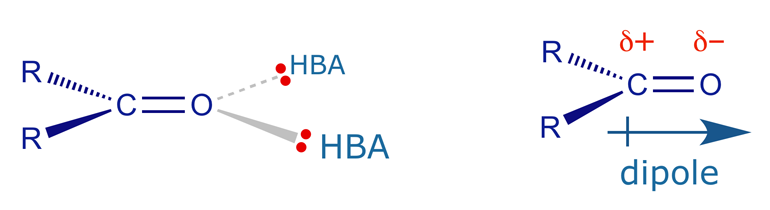
Ketone carbonyls are fairly common in bioactive structures. The group is planar and can interact with a binding site as an HBA through the two lone pairs of electrons on the carbonyl oxygen. The lone pairs are in sp2 hybridised orbitals which are in the same plane as the functional group. The carbonyl group also has a significant dipole moment and so a dipole-dipole interaction with the binding site is also possible.
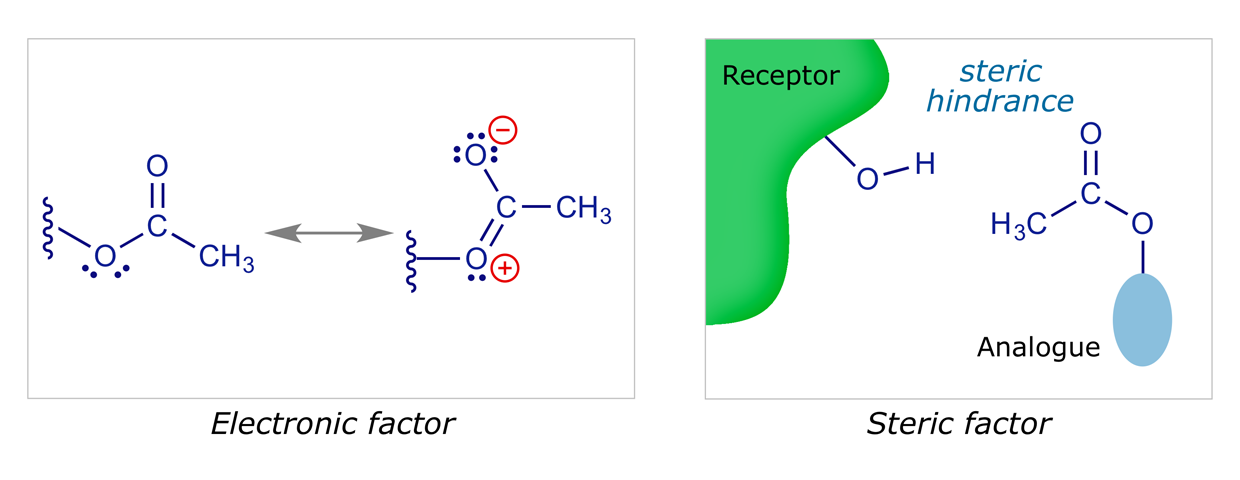
It is easy to reduce a ketone to an alcohol and it may be possible to carry out this reaction directly on the lead compound, changing the geometry of the functional group from planar to tetrahedral. Such a change in geometry may well weaken any existing H-bonding interactions and will certainly weaken any dipole-dipole interactions, as both the magnitude and orientation of the dipole moment will change. If it was suspected that the O-atom of the alcohol analogue might be acting as an HBA, the ether or ester analogues could be studied.
Amines are very important functional groups in medicinal chemistry and are present in many drugs. They may be involved in H-bonding either as an HBA or an HBD. The N-atom has one lone pair of electrons and can act as an HBA for one H-bond. Primary and secondary amines have N–H groups and can act as HBDs. Aromatic and heteroaromatic amines act only as HBDs, because the lone pair interacts with the aromatic or heteroaromatic ring.

In many cases, an amine may be protonated when it interacts with its target binding site, which means that it is ionised and cannot act as an HBA. However, it can still act as an HBD and will form a stronger H-bond than if it were not ionised. Alternatively, a strong ionic interaction may take place with a carboxylate ion in the binding site (right-hand graphic).

To test whether ionic or hydrogen bonding interactions are taking place, amines can be converted into amides. This will prevent the nitrogen acting as an HBA, as the nitrogen lone pair will delocalise into the carbonyl group instead. This interaction also prevents protonation of the N-atom and rules out the possibility of ionic interactions.

Amides are likely to interact with binding sites through hydrogen bonding. The carbonyl oxygen can act as an HBA and can form two hydrogen bonds. Both of the lone pairs involved are in sp2 hybridised orbitals which are located in the same plane as the amide group. The nitrogen cannot act as an HBA because the lone pair interacts with the neighbouring carbonyl group. Primary and secondary amides have an N–H group, which can act as an HBD.
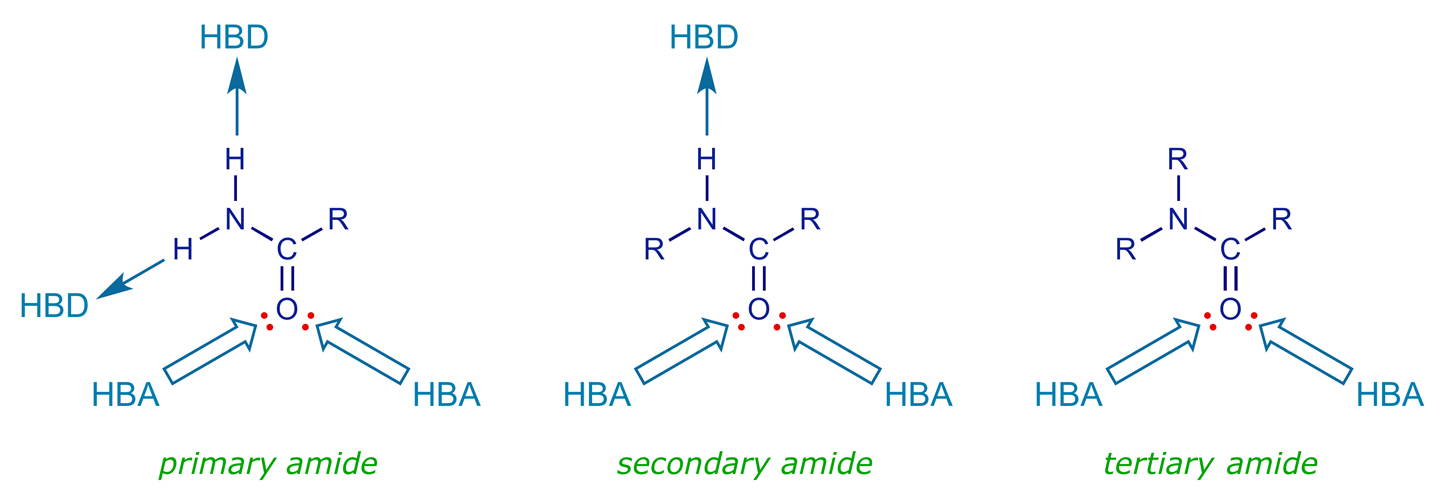
The most common type of amide in peptide lead compounds is the secondary amide. Various analogues can be prepared to test out possible binding interactions (see examples below). All of the analogues apart from the primary and secondary amines could be used to check whether the amide is acting as an HBD, and the alkenes and amines could be tested to see whether the amide is acting as an HBA (although the synthesis of some of these analogues may be difficult). It must be remembered that an amide group is planar, and does not rotate because of its partial double-bond character. The ketone, secondary amine and tertiary amine analogues have a single bond at the equivalent position, which can rotate (cf. captopril).

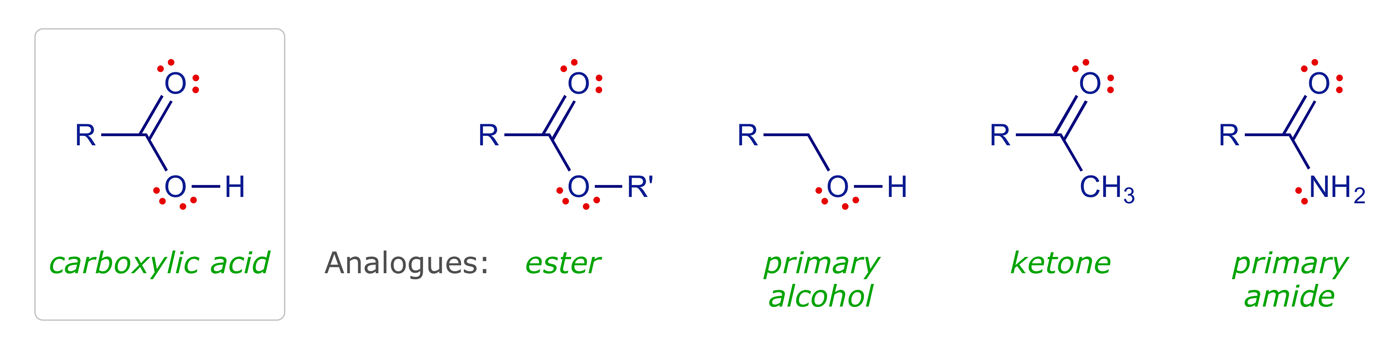
Carboxylic acids are quite common in drugs. The group can act as HBA or HBD, and may exist as the carboxylate ion, with ionic and/or strong hydrogen bonding with the carboxylate ion as an HBA.

To test for these interactions, analogues such as esters, primary amides, primary alcohols, and ketones are used. None of these functional groups can ionise, so a loss of activity could imply that an ionic bond is important. The primary alcohol could shed light on whether the carbonyl oxygen is involved in hydrogen bonding, whereas the ester and ketone could indicate whether the hydroxyl group of the carboxylic acid is involved in hydrogen bonding.
Ester groups have the potential to interact with a binding site as an HBA only. The carbonyl oxygen is more likely to act as the HBA than the alkoxy oxygen, as it is less hindered and has a greater electron density. The importance or otherwise of the carbonyl group could be judged by testing an equivalent ether. Esters are susceptible to hydrolysis in vivo by metabolic enzymes (esterases), which may pose a problem if the lead compound contains an ester that is important to binding, as it means the drug might have a short lifetime in vivo.

On the other hand, esters that are susceptible to metabolic hydrolysis are used deliberately to mask a polar functional group such as a carboxylic acid, alcohol or phenol so as to achieve better absorption from the gastrointestinal tract. Once in the blood supply, the ester is hydrolysed to release the drug. This is a common pro-drug strategy (see enalapril).
Heterocycles (cyclic structures with one or more of the heteroatoms O, N, or S) often feature in lead compounds, with N-containing rings particularly common. Heterocycles, which may be aliphatic or aromatic, have the potential to interact with binding sites through a variety of bonding forces. For example, the overall heterocycle can interact through van der Waals and hydrophobic interactions, while the individual heteroatoms present in the structure could interact by hydrogen bonding or ionic bonding.
With H-bonding there is an important directional aspect. The position of the heteroatom in the ring and the orientation of the ring in the binding site can be crucial in determining whether or not a good interaction takes place. For example, a purine ring can take part in six H-bonding interactions, three as an HBD and three as an HBA. Van der Waals interactions are also possible, to regions of the binding site above and below the plane of the ring system.

If the lead compound contains a heterocyclic ring, it is worth synthesising analogues containing an aromatic ring or different heterocyclic rings to explore whether all of the heteroatoms present are really necessary.
1.2 Isosteres and bioisosteres
Isosteres are atoms or groups of atoms which have the same valency (or number of outer shell electrons), and which have chemical or physical similarities. The groups SH, NH2, and CH3 are considered to be isosteres of OH, whereas S, NH, and CH2 are isosteres of O. Isosteres can be used to determine whether a group is important in binding, by altering the character of the molecule in a controlled way. For example, replacing O with CH2 makes little difference to the size of the analogue, but will have a marked effect on polarity, electronic distribution, and bonding. Replacing OH with the larger SH may not influence the electronic character, but steric factors may be more significant.

Isosteric groups have often been used to determine whether a particular group is involved in H-bonding. For example, replacing OH with CH3 would completely eliminate hydrogen bonding, whereas replacing OH with NH2 would not.
Bioisosteres are compounds or groups that possess near-equal molecular shapes and volumes, approximately the same distribution of electrons, and which exhibit similar physical properties. A bioisostere is the result of isosteric replacement with retention of the biological activity of interest. When replacin a group or atom in a lead structure with a bioisostere, some of the properties of that group will be retained, but various properties will be affected. The factors listed below are all important in determining whether a particular drug will get to the site of action and have the desired effect.
- Size
- Shape
- Electronic distribution
- Lipid solubility (hydrophobicity)
- Water solubility (hydrophilicity)
- pKa
- Chemical reactivity
- H-bonding ability
The application of bioisostere principles must be approached with caution. The examples listed below illustrate the fine line that may exist between a rational isosteric replacement and a mechanistically-sound bioisosteric replacement (in which the biological activity is retained).
Propranolol is a β-adrenergic blocker used to treat hypertension, anxiety and panic. The molecule has an aromatic ether linkage, and various analogues were made in which the OCH2 segment was replaced with the isosteres CH=CH, SCH2 or CH2CH2. These changes gave inactive compounds, but replacement with NHCH2 gave a bioisostere (i.e. the isostere retains the biological activity). These results confirmed that the ether oxygen atom is contributing to the activity of the drug, probably through H-bonding with the receptor.

Captopril emerged from an extensive study of analogues featuring isosteric replacement. The proline nitrogen atom is fundamentally important in controlling the shape of the molecule, as the C–N amide bond is rigid (partial double bond character). Clearly, replacing the N with the isosteric CH would not be a good move. The C–C bond would have no double bond character (C has no lone pair) and free rotation about the bond would be possible.

If a group or atom being replaced is involved in a specific interaction with a biological receptor or enzyme, then all the factors except lipid- and water-solubility will be important. Consider the consequences of replacing the thiol group of captopril. This group binds to a zinc ion in the active site of the target enzyme (ACE). If it is replaced, it must be with something that can also bind strongly to zinc ions. Replacing SH with CH3 (isosteric univalent groups in the table) would lead to a loss in binding ability, and hence poor activity.
Bioisosteric replacement represents a semi-systematic means of optimising the structure of a lead compound, but there are also more quantitative mathematical methods available — quantitative structure-activity relationships (QSARs).
2 Drug optimisation: Other strategies
The variation of lead structures in order to optimise their action (pharmacodynamics) and delivery (pharmacokinetics) can involve various strategies in addition to the conceptually simple business of identifying and 'tuning' the drug-target binding interactions. The following examples illustrate how some of the more subtle principles have been applied.
2.1 Structure extension
The strategy of extension involves adding another functional group to the lead structure so as to probe for extra binding interactions with the target. Lead compounds are capable of fitting the binding site and have the functional groups needed to interact with some of the important binding regions present. However, it is possible that they do not interact with all the binding regions available. For example, a lead compound may bind to three binding regions in the binding site but fail to use a fourth. Extra functional groups may locate that fourth region.
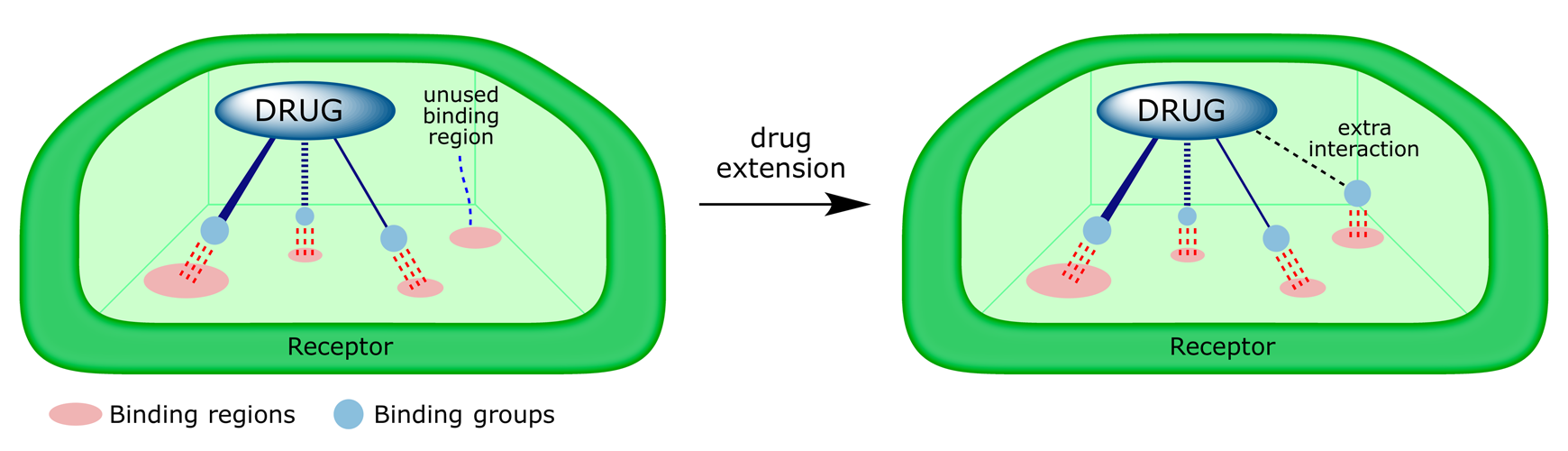
Extension tactics were used in the development of the second-generation ACE inhibitors in which the mercaptopropanoyl Zn-binder chain was replaced by a carboxylate chain. Adding a phenylalkyl group to the lead compound (I) increased activity (extension strategy).

Varying the length of the alkyl chain connecting the aromatic ring to (I) revealed that the best choice was a phenethyl group (chain extension strategy). The structure (enalaprilat) showed a 1000-fold improvement in inhibition, demonstrating that the extra aromatic ring was binding to a hydrophobic pocket in the enzyme's active site.
2.2 Variation of ring size
If a drug has one or more rings that are important to binding, a common strategy is to make analogues with the ring(s) expanded or contracted. The principle behind this approach is much the same as varying the substitution pattern of an aromatic ring, in that the positions of substituent groups are finely adjusted, and the angles slightly changed. Expanding or contracting a ring may put other rings in different positions relative to each other, and lead to better interactions with specific regions in the binding site.

This tactic was used during the development, by Roche, of the ACE inhibitor cilazaprilat (the corresponding pro-drug is cilazapril). The bicyclic structure I showed promising activity, the important binding groups being the two carboxylate groups and the amide group. By carrying out various ring contractions and expansions, cilazaprilat was identified as the structure having the optimum interaction with the ACE enzyme's binding site.
