
The Statins
- Cardiovascular disease
- Cholesterol
- The target enzyme: HMG-CoA reductase (HMGR)
- The discovery of the statins
- Type II statins
- Mechanism of action: Pharmacodynamics
- Mechanism of action: Binding of statins to HMGR
- Effect of statins on the liver
- X-Ray analysis of the HMGR binding site
1 Cardiovascular disease
The statins are a group of drugs that can reduce levels of cholesterol in the body by inhibiting the biosynthetic pathway that produces it. The market for cholesterol-lowering drugs is the largest in the pharmaceutical sector, and is dominated by the statins. For example, in 2002 the combined revenue of two leading drugs, atorvastatin (Lipitor®) and simvastatin (Zocor®), was more than USD 12 billion. Sales of atorvastatin since its approval in 1996 exceed USD 125 billion and it topped the list of largest-selling drugs for a decade. The patent expired in 2011 and, for a while, even the generics commanded a relatively high price.
The primary application of statins is for the treatment of dyslipidemia – an abnormal amount of lipids (e.g. cholesterol and/or fat) in the blood, often the result of diet and lifestyle choices. An elevated level of cholesterol is termed hypercholesterolemia, and a manifestation of this condition can be cardiovascular problems including coronary heart disease.
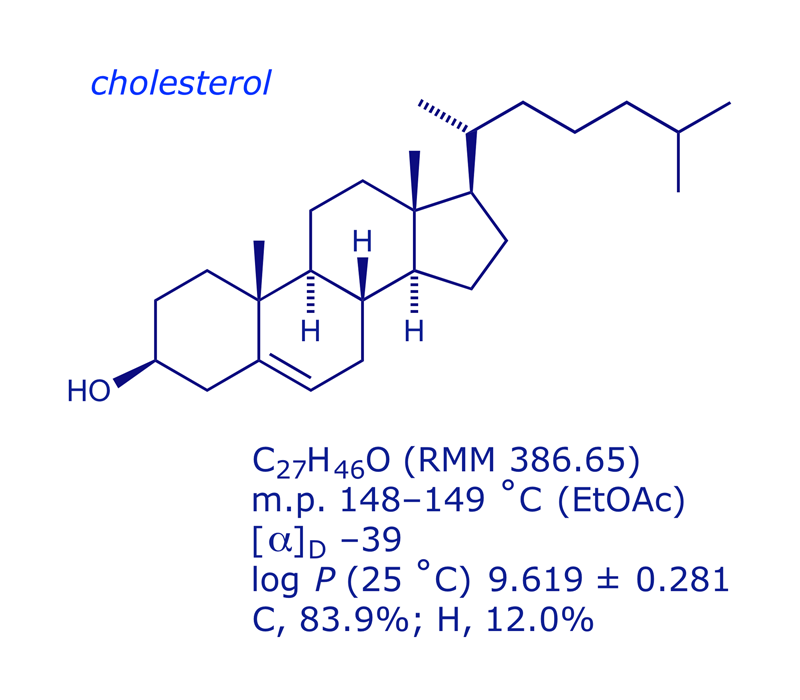
2 Cholesterol
Cholesterol is a major component of all biological membranes, and is the biosynthetic precursor of the steroid hormones, bile acids and vitamin D. Its presence is vital to healthy tissue and cells. It is biosynthesised by humans, and can be acquired in the diet.
What does cholesterol do?
- Major component of all biological membranes
- Biosynthetic precursor of steroid hormones
- Precursor of bile acids, vitamin D
Where does cholesterol come from?
- Biosynthesis in cells
- Consumed in the diet
What are the effects of too much?
- Hypercholesterolemia
- Cardiovascular disease
Cholesterol molecules in the membrane immobilise the first few hydrocarbon groups of the phospholipid bilayer. This makes the lipid bilayer less deformable and decreases its permeability to small water-soluble molecules. Cholesterol also prevents the crystallisation of hydrocarbons and limits phase shifts in the membrane. CLICK the image below to change the view.

The cholesterol molecule itself is almost all C and H, making it highly lipophilic. The transport of cholesterol through the bloodstream involves lipoprotein particles, of which there are two types. Low-density lipoprotein (LDL) particles are about 22 nm diameter and a molecular mass of about 3 000 000 Da. The protein (4536 AAs) encircles various types of lipophilic molecule (fatty acids, linoleic acid, phospholipids, cholesterol), keeping them soluble in blood plasma. LDL transports cholesterol and triglyceride lipids from the liver to other tissues. When a cell requires cholesterol, it produces LDL receptors which locate in the cell membrane; LDL binds to the receptors and is taken into the cell by endocytosis. High-density lipoprotein (HDL) particles are smaller (8–11 nm diameter), and transport fatty acids and cholesterol away from cells back to the liver, where they are removed from the blood. HDLs contain more protein than LDLs, and steadily increase in size as they pick up cholesterol on their way to the liver.
Coronary heart disease is associated with high levels of LDL ('bad cholesterol') and low levels of HDL ('good cholesterol'). If cholesterol from LDL is retained in the arteries, they become narrowed due to the formation of fatty plaques; atherosclerosis can result. Blocking of arteries supplying the heart (or brain) can induce a heart attack (or stroke). Statins are used to reduce cholesterol formation, and also cause an unanticipated lowering of LDL plasma levels.

3 The target enzyme: HMG-CoA reductase (HMGR)
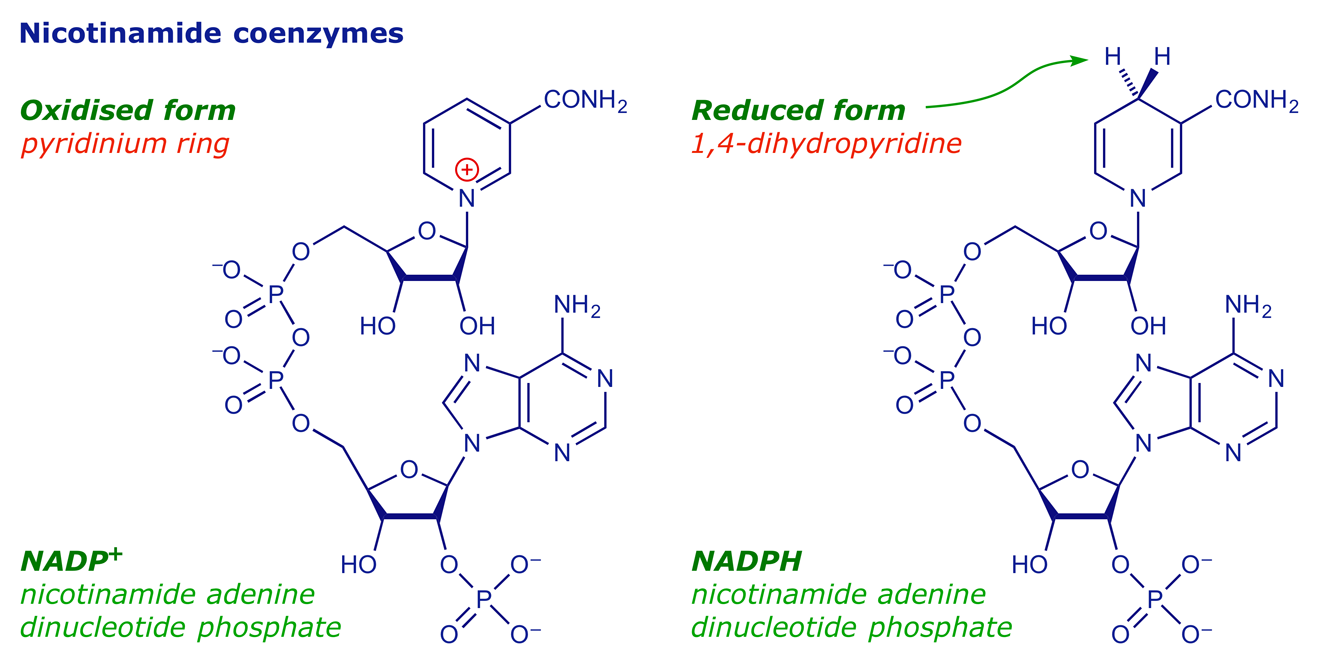
More than 30 enzymes are involved in cholesterol biosynthesis, and the pathway took decades to unravel. The enzyme that catalyses the rate-determining step of the whole process is HMG-CoA reductase (HMGR). The reaction involved is the conversion of 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) into (R)-mevalonic acid [(R)-3,5-dihydroxy-3-methylpentanoic acid], a reduction that requires two equivalents of the cofactor NADPH.


The HMGR enzyme (97 kDa) is a tetrameric membrane protein arranged as two dimers, with two active sites at monomer-monomer interfaces. The substrate HMG-CoA binds to one monomer, while the cofactor NADPH is located on the neighbouring monomer. The HMGR enzyme is highly flexible, and this was to prove important in the development of the statins.
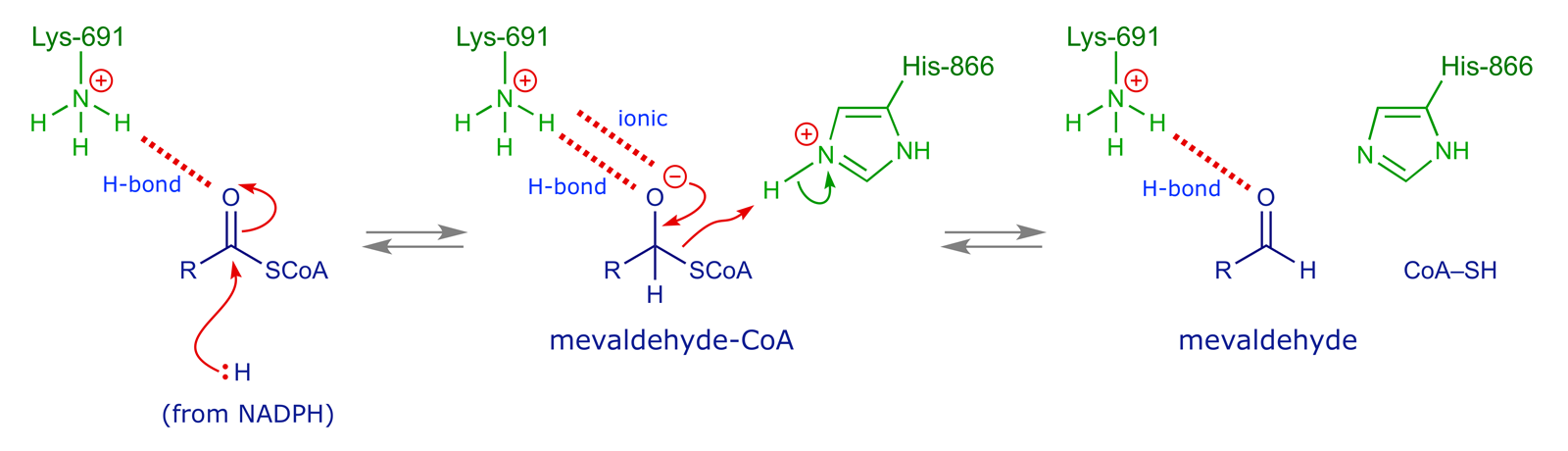
The reaction mediated by the enzyme involves two hydride transfers from the cofactor NADPH, of which the first cleaves off the CoA moiety and the second reduces the resulting aldehyde group.

It is known that in the substrate binding process, the carboxylate anion of HMG-CoA forms an ionic bond to Lys-735 of the enzyme. The substrate is also anchored by three hydrogen bonds: Ser-684 and Asp-690 cordinate to the 3-OH group, while the thioester oxygen atom interacts with Lys-691. The coenzyme A moiety is involved in various interactions with residues in a narrow hydrophobic slot close to the active site.

Other amino acids participate in the reaction mechanism. A histidine residue (His-866) acts as an acid catalyst, supplying the proton required for the CoA to become a leaving group, while the coordination of the thioester oxygen to Lys-691 lowers the activation energy for the first hydride reduction step by supporting the build-up of negative charge on the O atom.
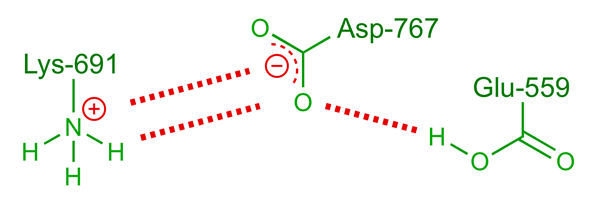
An uncharged glutamic acid, Glu-559, is also involved in the reaction, providing the proton that quenches the second reduction step in which mevaldehyde is reduced to mevalonate. The lack of a charge on Glu-559 is unusual — in this case the situation is brought about by the proximity of Asp-767, which serves to stabilise the protonated forms of both Glu-559 and Lys-691 through the hydrogen-bond network illustrated below.

4 The discovery of the statins
Once HMGR was identified as a potential target, the search began to find an inhibitor, and screening of the natural products produced by microorganisms provided a lead, compactin.

Compactin was isolated from Penicillium citrinum in the 1970s. It was identified as a potent HMGR inhibitor (10000-fold higher affinity for the enzyme than the natural substrate). In 1978 Merck isolated mevinolin, an analogue of compactin (differing by one methyl group), produced by Aspergillus terreus. Clinical trials of mevinolin began in 1980 and it was eventually marketed as lovastatin, a landmark in the treatment of hypercholesterolemia.
Other statins soon followed. Simvastatin, approved in 1988, was prepared by semi-synthesis from lovastatin. Pravastatin was obtained from compactin by biotransformation and first marketed in 1991. These are type I statins, all ultimately derived from fungal metabolites and having the general structure shown below.

It should be noted that, while lovastatin and simvastatin lack the polar 'head' group shown in the generalised structure, these compounds are both pro-drugs. Their respective lactone rings are hydrolysed in vivo to produce the corresponding hydroxy acid form.
5 Type II statins
The type I statins proved to be very effective at lowering cholesterol levels, but the associated side-effects and difficult syntheses prompted further optimisation work. This yielded the so-called type II statins, in which the decalin ring is replaced by a larger hydrophobic moiety.
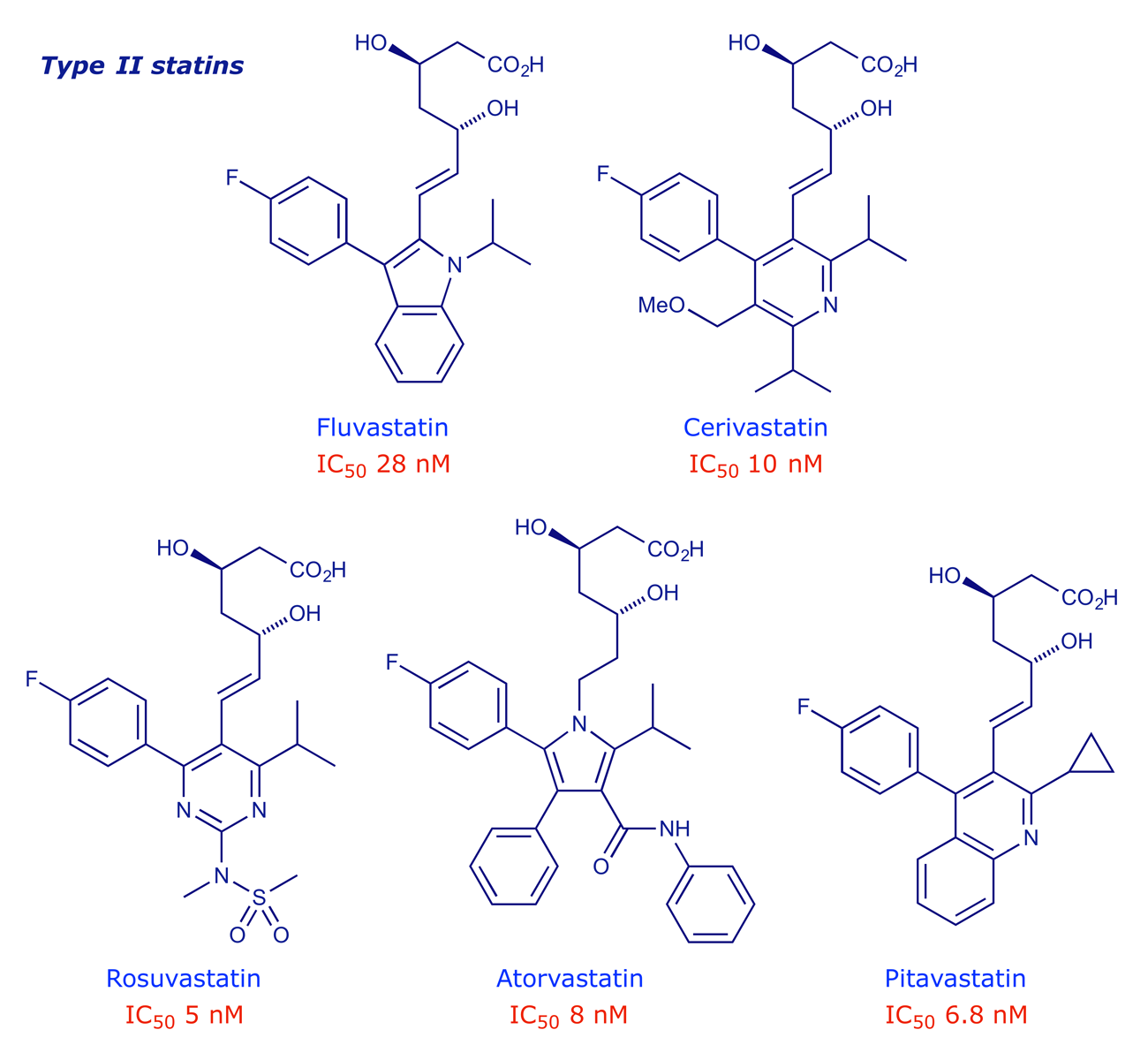
Type II statins are much easier to synthesise as they lack all but two of the stereogenic centres present in the type I series. They can be regarded as 'me-too' and 'me-better' drugs, i.e. their designs are strongly influenced by what went before, but left room for intellectual property rights to be securable. PK studies have revealed a spectrum of effectiveness that depends, at least in part, on the hydrophobic nature of the molecule. Lower hydrophobicity makes a statin more selective for liver cells, where most of the cholesterol biosynthesis takes place.

Rosuvastatin is the most potent of the structures shown above. The sulfonamide group was added for the specific purpose of making a drug that was less hydrophobic.
6 Mechanism of action: Pharmacodynamics
The statins are competitive inhibitors of HMGR. They mimic the natural substrate, competing with it for the active site on the enzyme — in fact they bind more strongly. The core structure of the statins incorporates a polar head group that mimics the natural substrate, together with an extensive hydrophobic group that can bind more strongly with the hydrophobic binding region of the enzyme.
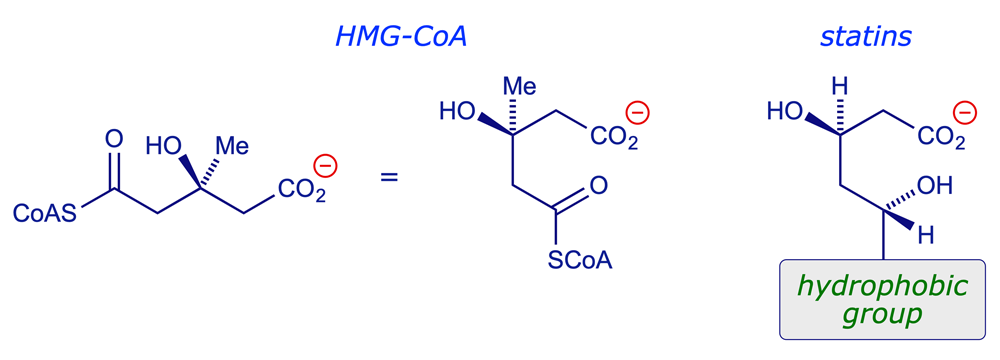
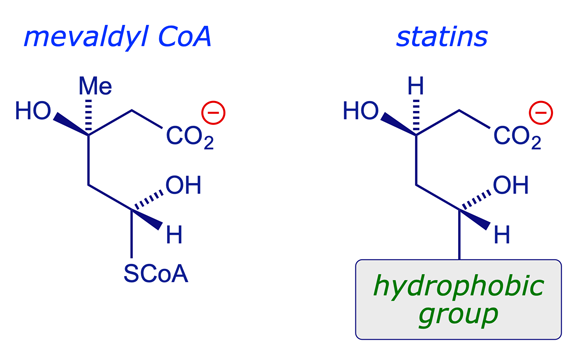
7 Mechanism of action: Binding of statins to HMGR
The binding of HMG-CoA and various statins to the HMGR enzyme has been studied by X-ray crystallography, revealing the nature of the enzyme binding site and the atoms/groups that interact directly with the substrate. The key interactions are shown in the diagrams below, and can be confirmed by inspecting the X-ray structures themselves (see Section 9).
An important conclusion that emerges from the X-ray analysis runs counter to any suggestion that the hydrophobic region of the statin occupies the same part of the HMGR enzyme as the coenzyme A unit of the natural substrate. It transpired that the CoA binding pocket is too narrow for this to be the case. Nor is there any other hydrophobic region that could accommodate the side-chain. For these reasons it is concluded that the enzyme possesses significant flexibility. When HMG-CoA substrate binds, an α-helical section of the protein folds over the active site, shielding it from water and creating a narrow hydrophobic cleft that accepts the CoA section of the molecule. When a statin binds, the enzyme alters its shape (induced fit) differently, with the formation of a shallow hydrophobic binding region next to the active site as a result of the movement of two C-terminal α-helices (Lα10 and Lα1) whose flexibility is a key factor.
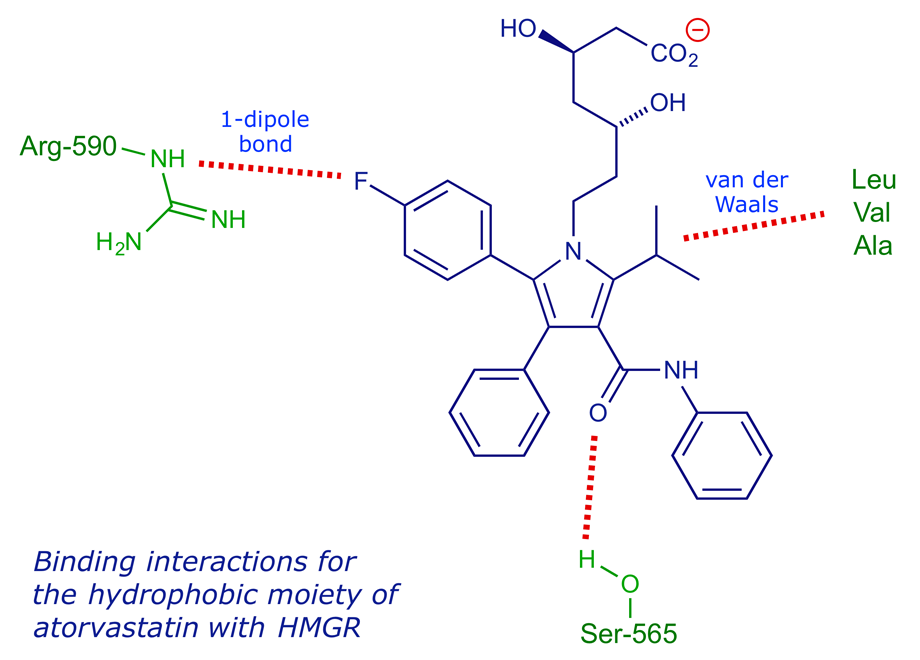
In the binding of type II statins, the isopropyl group present in most examples binds to the same part of the narrow hydrophobic region as the decalin ring of the type I systems, but there are extra interactions (van der Waals type) with hydrophobic side-chains in this locality. The fluorophenyl moiety can coordinate to Arg-590 of the enzyme through dipolar and π-stacking interactions. With atorvastatin, the carbonyl group of the amide group provides an additional H-bonding locus, while rosuvastatin can do the same via its sulfone group.
8 Effect of statins on the liver
The action of statins is not solely due to their inhibition of HMGR. In fact, the biological response to statin intervention includes the upregulation of HMGR synthesis, as a means of counteracting the inhibition. Another effect was discovered after the introduction of statins: Lowering of the level of cholesterol in the liver causes an increase in the production of liver LDL receptors, and these increase the capacity of the liver to clear LDL-cholesterol from the plasma. This is a key factor in the effectiveness of statins in controlling cholesterol levels.
9 X-Ray analysis of the HMGR binding site
Interactive 3-D molecular models of HMGR complexed with HMG-CoA (the natural substrate) and HMGR complexed with atorvastatin (Lipitor®) are provided. They are based on the X-ray analyses published by Deisenhofer and coworkers in 2000 and 2001 respectively.
Crystal structure of the catalytic portion of human HMG-CoA reductase: insights into regulation of activity and catalysis
E. S. Istvan, M. Palnitkar, S. K. Buchanan and J. Deisenhofer, EMBO J., 2000, 19, 819–830.
Structural mechanism for statin inhibition of HMG-CoA reductase
E. S. Istvan and J. Deisenhofer, Science, 2001, 292, 1160–1164.
Interactions between the HMG moiety of atorvastatin and the protein are mostly ionic or polar. The rigid hydrophobic groups of atorvastatin are situated in a groove between helices Lα1 and Lα10, and there are additional interactions between Arg-590 and the fluorophenyl group (the guanidinium group of Arg-590 stacks on the fluorophenyl group, with polar interactions between the arginine nitrogen atoms and the fluorine atom). There is a hydrogen bond between Ser-565 and a carbonyl oxygen atom.
Atorvastatin (Lipitor)
