
Principles and Terminology
1 A brief history of 'MedChem'
The search for cures to human ailments extends throughout human history, and it is instructive to look at the developments over the centuries and see where we are in the scheme of things. It is accepted that the current state of human health has been about 4.5 billion years in the making, and the first life forms, e.g. cyanobacteria, are thought to have prompted the change in the earth's atmosphere from reducing to oxidizing over about two billion years, stimulating biodiversity.

If the scale of this timeline were to be scaled to one year, then the emergence of man would be on 31 December at about 23:40, while agriculture was introduced in the final few minutes before midnight. Medicinal chemistry is brand new technology.
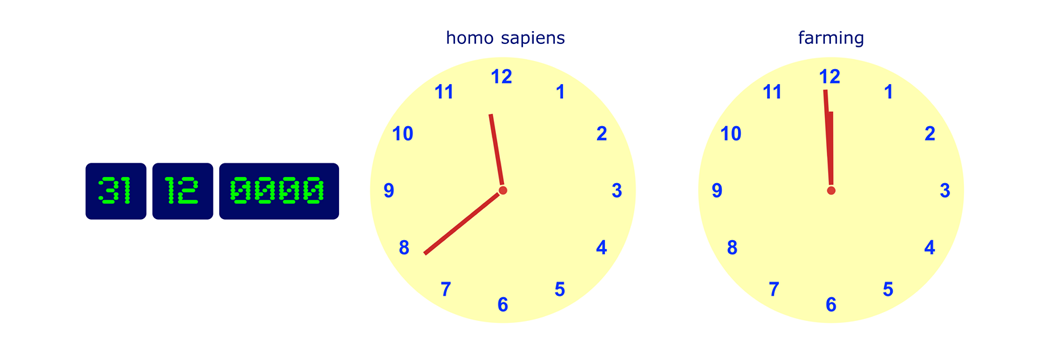
Man has attempted to control disease and ailments since he came into existence. The earliest known 'treatments' are reported on a Sumerian clay tablet dated ca. 2100 BC, but this record does not identify any ailments. More detailed records are given in the Ebers papyrus from Ancient Egypt (ca. 1500 BC), which lists (in hieratic Egyptian) about 700 recipes and their uses. The treatments were crude multi-component preparations made from plants, minerals, and animals, and were often used in association with religious rituals and incantations.
Some of these ancient treatments were refined over the following centuries, but it was not until the nineteenth century, and the growth of organic chemistry as a science, that pure biologically active components of these mixtures were isolated and some understanding emerged of how they worked. Many important milestones have been passed in the last 200 years (see Appendix 1: Some Important Events in Medicinal Chemistry), including:
- Isolation of morphine and its commercial scale production (1800–1833): The first medicinally useful material to be isolated in a pure form on a commercial scale was morphine, a pain control that is still used today. The large scale production of morphine from poppies was first undertaken by Macfarlane & Co., Edinburgh, and this probably represents the start of the modern pharmaceutical industry in the UK.
- Anaesthetics (1840s): The introduction of ether, nitrous oxide and chloroform as anaesthetics transformed surgery. It was no longer necessary to perform a surgical operation at speed, and precision became possible. Survival rates after surgery increased dramatically (see also the use of phenol as an antiseptic, 1867).
- First synthetic drug (1897): Erlich introduced salvarsan, the first synthetic drug, for the treatment of trypanosomiasis (sleeping sickness). This represented a significant departure from the earlier practice of only using materials isolated from nature.
 ×
× - The isolation and limited-scale production of penicillin (1929, 1940): Death due to bacterial infection was commonplace until penicillin became available. Its discovery ushered in the development of an arsenal of antibacterial drugs that have contributed greatly to the improved life expectancy and quality in recent times.
- Rapid growth of the pharmaceutical industry (1960–2000): Many advances were made in medicinal chemistry and biology during these decades and a wide range of drugs were developed. In particular, our understanding of heart disease, cancer, viral and fungal infections is more extensive and 'smart' therapies have emerged.
Following the spectacular emergence of penicillin from a mould, all drug companies realised that Nature is billions of years ahead of us when it comes to the synthesis of biologically active compounds. Many new natural products were isolated, identified, modified, and screened for biological effects. Research methods varied between companies, reflecting differences in local expertise, but drug discovery grew up as a free-standing technology in its own right. The suggestion, by Erlich in 1908, that a disease might be cured by dosing the patient with a compound that acted as a magic bullet has been vindicated.
Key definitions and central issues are summarised below (for a full list, see Appendix 2: Glossary of Medicinal Chemistry Terminology).
KEY DEFINITIONS
| Drug | A compound that interacts with a biological system to produce a biological response. |
|---|---|
| Safety | No drug is totally safe. Side-effects vary and can include death. |
| Dosage | The dose level of a drug determines whether it will be beneficial or a poison. |
| Therapeutic Index | The ratio of the dose providing a beneficial effect versus the dose that is harmful. A high therapeutic index means a large safety margin. |
| Selectivity | Ideally a drug should target the abnormal medical condition without adversely affecting anything else. |
2 The drug discovery pipeline
Drug discovery is a hunt for new or better agents for tackling human diseases. In general, discovery programmes follow a sequence resembling the one outlined below, in which two entry points are specified. Some of the stages run concurrently and involve considerable overlap: clearly the feedback from testing must inform the design process. Chemists are heavily involved at all stages until the clinical trials. Their tasks are (i) to identify new leads, (ii) to optimise leads to provide clinical candidates, and (iii) to develop large-scale syntheses.
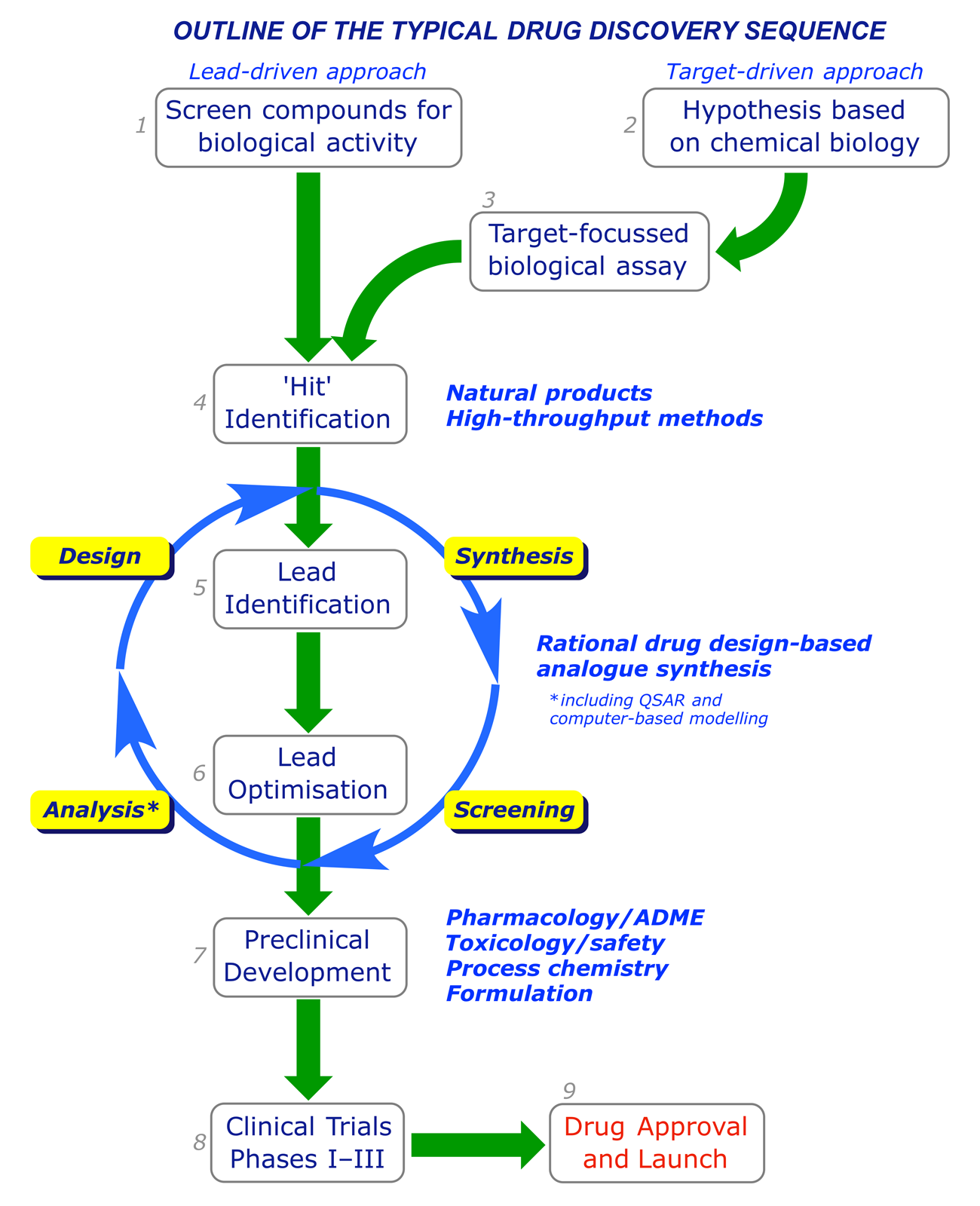
2.1 The lead-driven and target-driven approaches
Traditionally drug discovery has been lead-driven. Natural or synthetic compounds (pure or as mixtures) from any source were screened for their effects on particular microorganisms (e.g. bacteria), cell lines, enzymes or tissue types. After exposure to a test compound, cells or microorganisms can be checked for viability using staining methods. Ligand-receptor or ligand-enzyme interactions can be quantified using radioligand competition techniques. The progress-limiting factor of this approach is throughput, and in recent times the emphasis has been on testing as many compounds as possible. High-throughput technologies (combinatorial chemistry and screening), made possible by advances in digital control methods and robotics, allow rapid searching for 'hit' compounds using automated synthesis and screening equipment.
Advances in biotechnology have led to drug discovery becoming more target-driven. Interdisciplinary teams with expertise in genomics, structural biology, biological chemistry, computational modelling etc., compare healthy and diseased states, build a complete picture of the molecular mechanisms involved, and look for a strategic target, e.g. one enzyme in a cascade. Potential therapeutics are rationally designed on the basis of the properties (shape, functional group array, log P etc.) considered most likely to assist their hitting the target.
Lead-driven approach
- Trial and error
- High-throughput technologies
- Separate chemistry and biology
- Low degree of chemical specialisation
Target-driven approach
- Focus on selected biological targets
- Rationalise chemical and biological data
- Interdisciplinary teamwork
- Greater specialisation, new skills
2.2 Target-focussed biological assay
The hypothesis resulting from a target-driven analysis guides the choice of bioassay required for identifying 'hit' compounds. Establishing a bioassay typically involves the following:
Setting up a focussed biological assay
- Find an organism that can express the target protein
- Grow a batch of the protein, isolate and purify it
- Characterise (structure, properties, enzymic activity etc.)
- Crystallise the protein and obtain its X-ray structure
- Develop an assay based on the protein's enzymic activity
The 'organism' required for this task is often a strain of bacteria or yeast that is known — or has been engineered — to express (metabolically produce, biosynthesise) the target protein. The eventual assay may involve the measurement of the action of the target protein directly, or measuring some effect elsewhere (downstream) that its activity causes to happen.
A biological assay to be used for compound screening must be robust and give reproducible results. Ideally the method will also be amenable to validation with available standards ('tool' compounds); work in the presence of non-aqueous solvents such as DMSO; compatible with medium/high thoughput automated assay systems. Some chemical synthesis may be required to develop bioassay tools, e.g. if suitable calibration standards are not commercially available.
2.3 Hit identification
With a biological assay in place, the search for hit compounds can begin. A hit could be defined as a compound that meets preconceived threshold criteria for 'activity' in the primary screening assay. Materials that are screened include natural products, synthetic compounds from any source, and custom-made combinatorial 'libraries.' The choice of materials for screening can be random, but may be limited or targeted on the basis of theory or experience. Very few hit compounds progress further to become leads (see next section), but any structure-activity data might be useful in defining the structural requirements for activity.
2.4 Lead identification
A lead is defined as a compound that is reproducibly a 'hit', whose structure is deemed to be modifiable into analogues with 'drug-like' physicochemical properties. These materials will meet the target (rather than threshold) criteria of the primary biological assay, but may still not be suitable for pharmaceutical use — they may be weakly active, unstable, too expensive to produce, have off-target effects or be unpatentable, but they can still guide chemists to a final drug structure. The following criteria are established in identifying lead compounds:
- Potency: A hit compound is initially re-tested under the original assay conditions. Then its potency is quantified using a dose response curve, generated by measuring the effect of the compound at different concentrations. The potency is quantified in the IC50 or EC50 value.
 ×
× - Efficacy: Activity will be measured in other assays. At least one of these will be cell-based, to confirm that the compound can pass through biological membranes on its way to the target.
- Tractability: Medicinal chemists will assess whether the hit structure is amenable to chemical modification, i.e. that analogues with more drug-like properties will be accessible.
- Availability: A hit compound is assessed for ease of synthesis at a reasonable cost, bearing in mind the eventual requirement for large-scale production.
- Patentability: The intellectual property (IP) rights are verified using appropriate databases.
2.5 Lead optimisation
Once a compound with the targeted activity meets the 'lead' requirements, the focus shifts to improving its pharmacological profile, or drug-like properties. Guided by established design strategies (discussed later), medicinal chemists synthesise modified structures and the assessment process is reapplied, in collaboration with the pharmacology team.
Issues addressed by pharmacologists
- Does the biological activity transfer to animal models?
- Is the compound toxic?
- Is the compound absorbed by tissues other than its target?
- Does the compound possess oral bioavailability?
- How quickly/slowly is the compound metabolised?
Many promising compounds fail to provide good answers to such questions. The potency of a compound is relatively easy to manipulate, but predicting or changing its pharmacokinetic profile is a different matter. The relevant issues are referred to as ADME:
- Absorption: Experiments in vitro can be used to measure how well a compound is absorbed by cells, and the results provide some predictive power. However, in vivo (animal) studies must be used to analyse absorption characteristics in depth.
- Distribution: Once absorbed, a compound is distributed around the body. The distribution characteristics will depend on the formulation and chemistry of the drug, as well as the route of administration. Animal studies are used to analyse a drug's distribution characteristics.
- Metabolism: Compounds are metabolised or modified biologically as the body goes through the process of clearing them. The liver is the most significant metabolic organ, but other sites occur throughout the body. The metabolites generated may be active or inactive in terms of pharmacology. In lead optimisation, basic in vitro and in vivo studies are performed to analyse metabolic events and see how the body deals with the drug.
- Excretion: Drugs and their metabolites can be eliminated from the body by various routes, e.g. in the urine or faeces, by exhalation etc. Animal studies are used to analyse the excretion characteristics of a drug candidate molecule.
There are tools, e.g. Lipinski's Rule of five, that support these aspects of the lead optimisation phase and can guide drug designers on the issue of absorption:
Lipinski's RULE OF FIVE
A compound is more likely to be membrane-permeable and easily absorbed by the body if it matches the following criteria:
- The molecular mass is below 500
- The lipophilicity, expressed as the octanol-water distribution coefficient log P, is less than 5
- The substance has less than 5 hydrogen-bond donor groups
- The substance has less than 10 hydrogen-bond acceptor groups
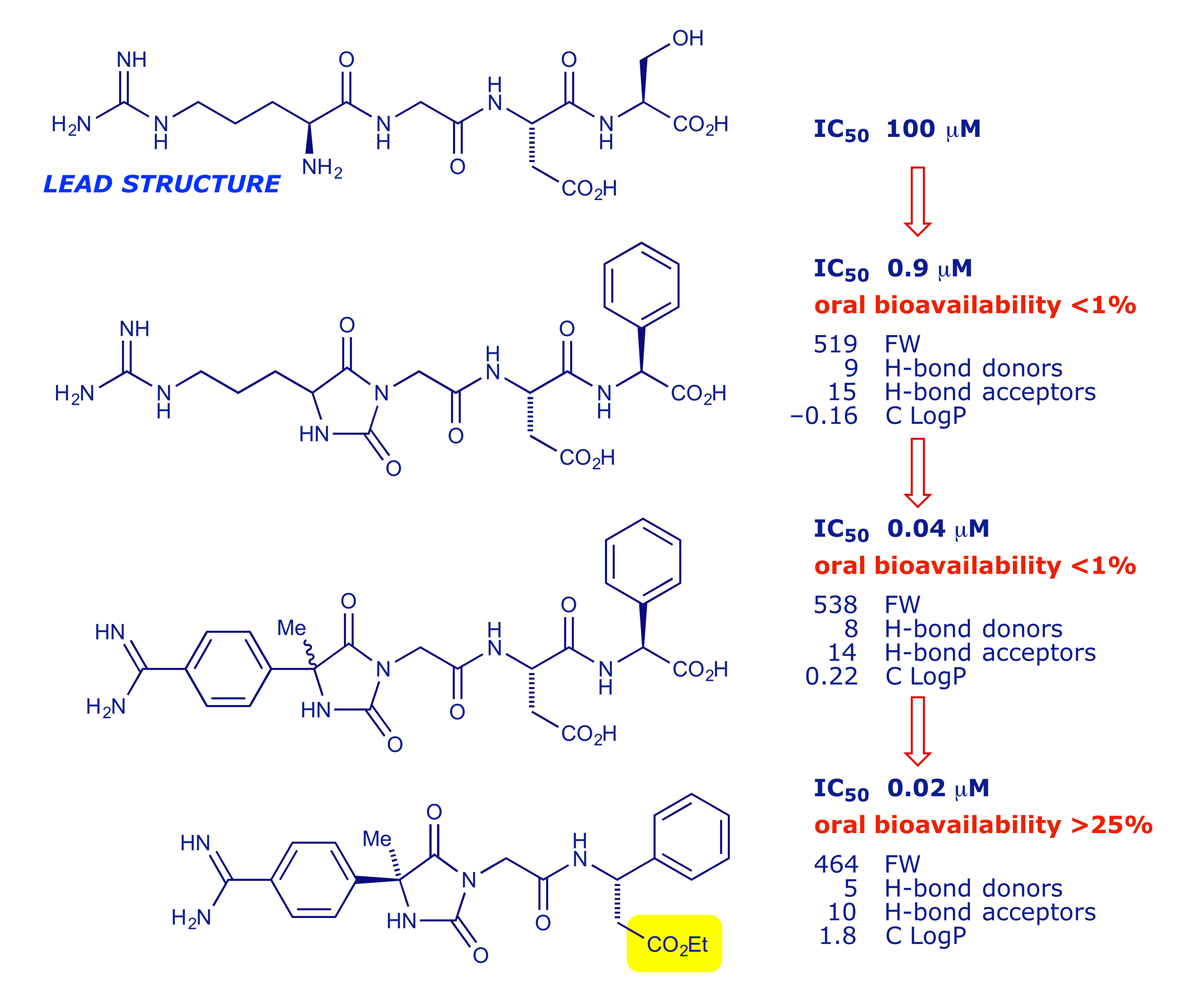
The value of the Lipinski analysis is illustrated below. The peptide RGDS was a lead structure in a project targeting fibrinogen-receptor antagonists for use as antithrombotics, which are potentially valuable for treating or preventing heart and circulatory problems. The biological activity of the lead structure was quickly improved, but 'bioavailability' remained an issue until the carboxylic acid group was esterified (read the paper here).
2.6 Preclinical development
Pharmacologists study in detail how drug candidates interact with a molecular target, using in vitro and in vivo assays to quantify receptor binding, inhibition, kinetics, efficacy, potency, dose responses, duration of effect etc. The pharmacology has two distinct categories:
Pharmacokinetics (PK)
- The fate of a drug molecule, i.e. what happens to it once inside the body. This is the sum of the ADME processes.
Pharmacodynamics (PD)
- The physiological effect of a drug molecule, i.e. what it causes to happen once inside the body.
Human trials must always be preceded by safety testing in animals. The majority of potent drugs and compounds will have side-effects that could be dose-limiting as far as human use is concerned. Toxicology is the study of the potentially toxic and unwanted effects of drugs. In lead optimisation, various in vitro and in vivo studies are used to identify the dose-limiting effects of compounds, the aim being to identify candidates with the optimal therapeutic index (efficacy v. side-effects). There are strict guidelines as to which species should be used in preclinical toxicology to ensure that the results can be used to predict effects in humans.
Once a suitable candidate has been identified, it is required in large quantities. Material used and tested during lead optimisation is often prepared only in milligram quantities, but clinical trials require kilograms. Process chemistry is the science of using an in-depth knowledge of chemical reactions to develop a robust, large-scale and economic synthesis that can be used to make multi-kilograms of a drug candidate.
At a late stage, a drug candidate is submitted for formulation studies in which the objective is to ensure that the drug is presented, for clinical use, in a suitable form (size, taste etc.) for administration to a patient. Ideally the drug will also be formulated for a long shelf-life in a pharmacy (or home), remaining stable and unaffected by light, heat or moisture.
2.7 Clinical trials
Testing a new drug in humans is a highly regulated process consisting of controlled sequence of closely monitored clinical trials, referred to as Phases I–III. Should a drug reach the market, its effects on patients will be closely monitored by the company (Phase IV).
- Phase I (about 1 year): Only about 1 in every 2500 compounds tested get to this stage, and many compounds that enter Phase I trials will fail to make it to the market as drugs. The compound is administered to 50–200 normal healthy volunteers, with the main objective of determining if any severe side-effects are present. At this stage it is also established what dose range should be used, and some investigation into how the drug is absorbed, metabolised and excreted is undertaken.
- Phase II (about 2 years): Once the drug has been successfully tested in healthy volunteers it can be submitted for Phase II clinical trials. This involves testing the drug in patients with the target disease. These trials usually involve 100–500 volunteers who suffer from the target illness, and the patients are divided into two groups: one group is given the prospective drug, and the other group is given a placebo (an inactive substance resembling the drug). To ensure that the trials are unbiased neither the patients nor the doctors administering the treatment know whether the drug or the placebo is being used (double-blind trial). Phase II trials take about two years to complete, and provided that the drug performs better that the placebo and no longer-term side-effects are observed then it can proceed to the next stage.
- Phase III (about 3 years): Phase III trials involve considerably more patients (1000–5000) and can take up to 3 years to complete. These trials usually take place all over the country, and in some instances are conducted on an international basis. It is in this phase that the beneficial effects, or otherwise, of a drug are finally proven and evaluated. Rare but significant side-effects can also come to light in Phase III.
2.8 The cost of drug development
The timeline and cost of drug development are the subject of intense debate among the various stakeholders. All agree that the investment required is enormous, but the estimates of the true cost range from $0.5–2 billion. One of the most cited estimates, published in 2003 by DiMasi et al., put the capitalised cost of taking a drug to the clinic at $802 million at year 2000 values, and the time/cost chart below is guided by DiMasi's total.
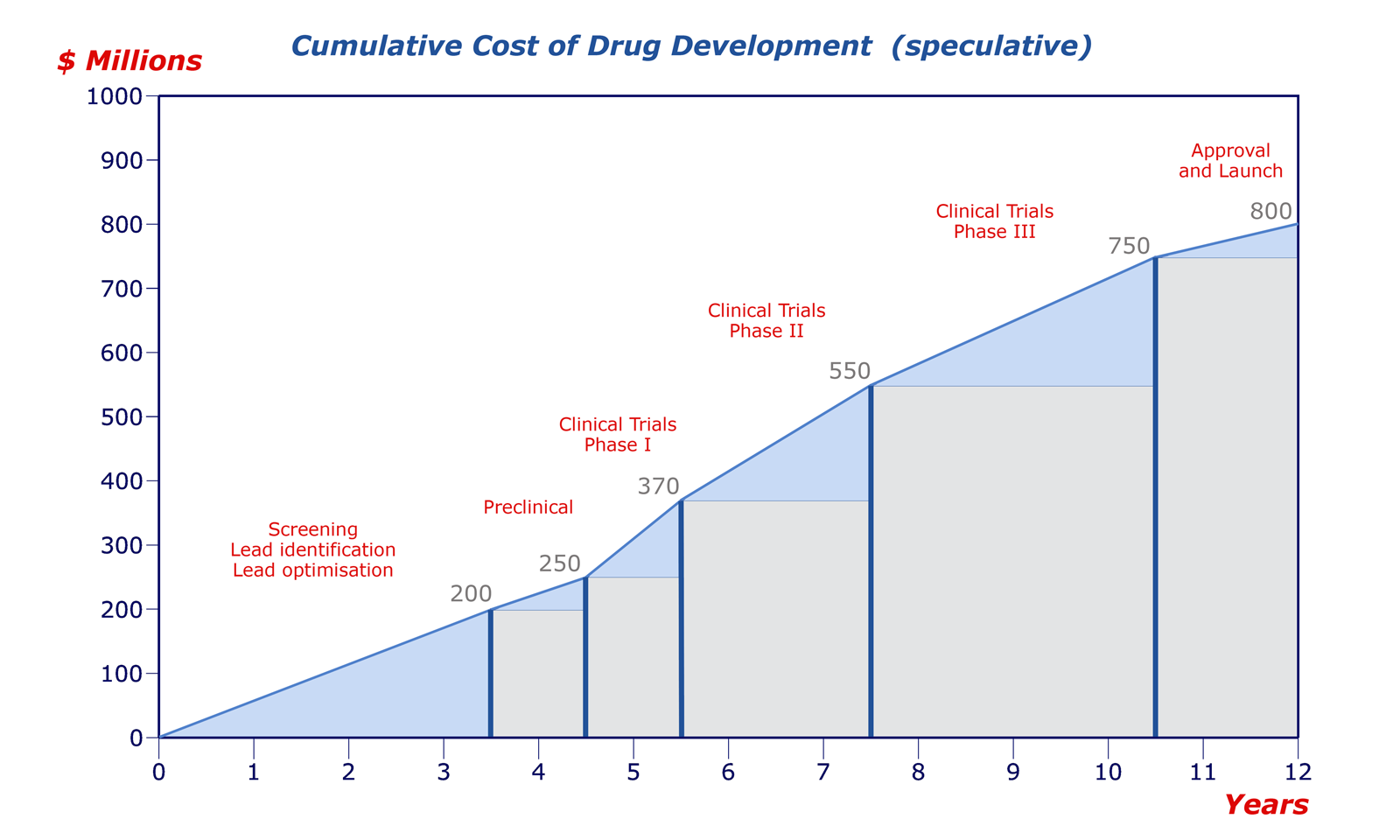
The positions of the divisions are speculative, but it is accepted that the most costly part of the sequence is the clinical trials. From a strategic point of view, the perceived wisdom is therefore 'fail early, fail cheap.' Clearly, a company does not want to discover a major problem after ten years of effort and £500 million of investment, late in clinical trials or, in the worst case, after a drug has been marketed. Late-stage toxicity failures are all too common, and there is often a strong case for bringing forward ADME studies.
Some of the published responses to the costings by DiMasi et al. are listed below.
• Extraordinary claims require extraordinary evidence
• Estimating the cost of new drug development: Is it really $802 million?
• Drug development cost estimates hard to swallow
• How Big Pharma distorts the costs of developing new drugs
3 Selecting drug targets
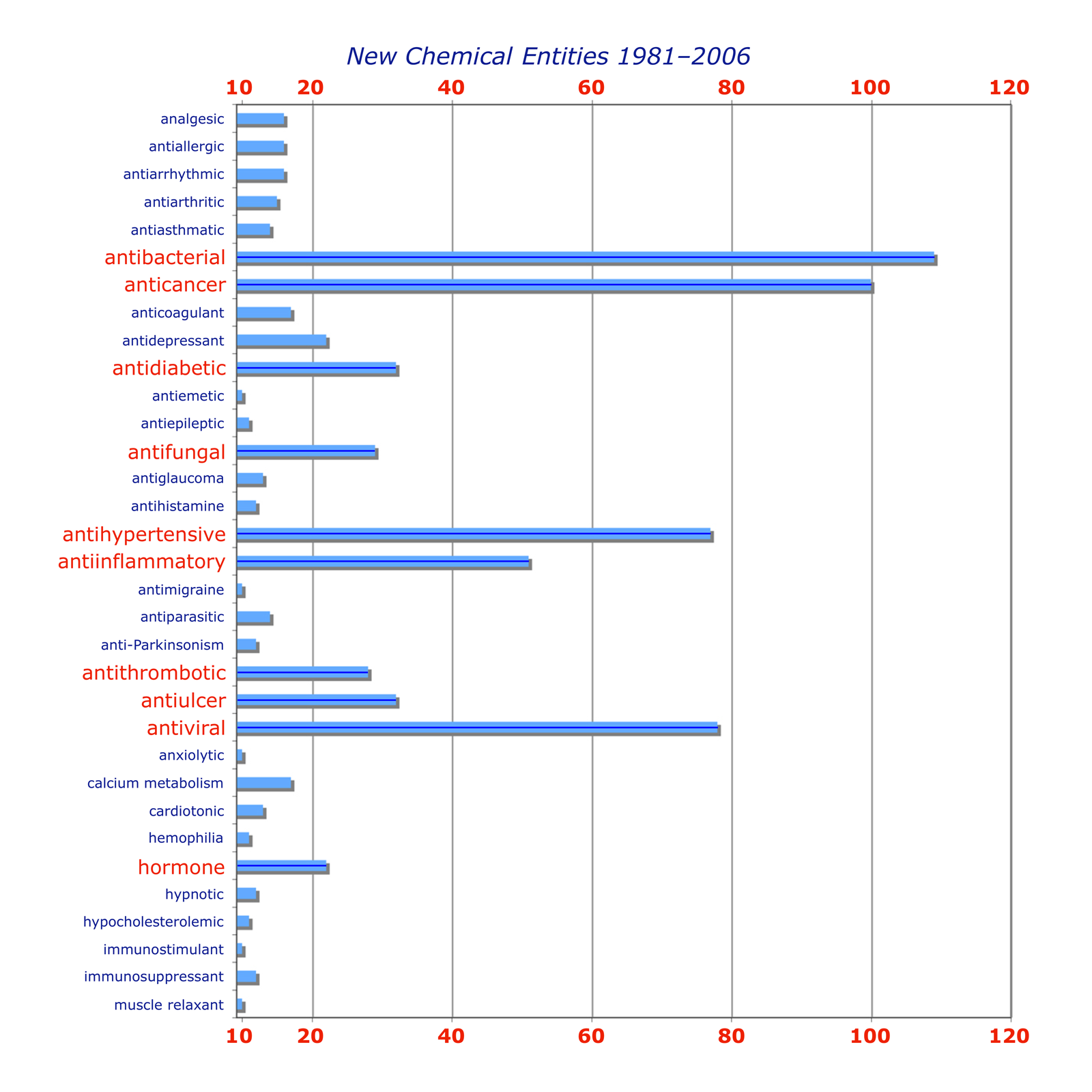
The selection of a target by a drug company or research organisation — the decision to seek an agent with a biological effect that has therapeutic utility — involves many stakeholders and takes in a range of scientific, medical, financial and strategic considerations. For a detailed analysis, see this review: J. Knowles and G. Gromo, Nat. Rev. Drug Discovery, 2003, 2, 63–69 (doi: 10.1038/nrd986). The chart below, taken from the review, serves to illustrate the potential complexity of the decision-making process. We will focus on the science involved.
3.1 Choosing a disease
The primary objective in drug discovery is compounds to be used as a treatment for illness or pain, and obviously there is no shortage of targets. The major drug companies generally distribute their efforts between several of broad areas (oncology, central nervous system, infections etc.). Commercial drug research often targets ailments that are prevalent in the 'developed' world (heart disease, cancer, ulcers, migraine etc.), while the fight against malaria, one of the world's most lethal infectious diseases, has been taken up over the years by organisations such as the World Health Organisation, The Gates Foundation and the US Army, for whom financial gain was not the primary concern. (What was? You decide.)
3.2 Choosing a drug target
Identifying a medical need is relatively straightforward, but organising a biological effect that will alleviate the problem is not easy. Many early drugs were based on natural products that were found to have a biological effect, discovered through random screening. How they worked often remained obscure for years, but recent developments provide a new situation:
Most drugs target proteins
- Enzymes
- Receptors
- Ion channels
The structures of proteins are known
- Genome projects have mapped human DNA
- Other species are similarly encoded
- The role of many proteins remains unknown
Some drugs target nucleic acids
- DNA
- RNA
Choosing a drug target has increasingly become a matter of identifying the proteins that are involved in the targeted condition, and designing a molecule that will interact with them. The issue of selectivity remains crucial if side-effects are to be minimised or avoided.
Selectivity between species can be relatively easy to achieve. For example, penicillin targets an enzyme that mediates cell wall biosynthesis in the bacteria that cause the infection. Mammalian cells do not have a cell wall, so knocking out this enzyme does not affect humans. On the other hand, some infectious organisms have enzymes that are similar, but slightly different, to a human equivalent, and the differences can be targeted.
Selectivity within the body is also desirable. An enzyme inhibitor should only target one enzyme, and receptor agonists/antagonists should only target one type of receptor. But bear in mind that more complex issues might lie in wait — the body has a highly interactive system of messengers, enzymes and receptors, and the imbalance caused by a drug can bring about unexpected (and undesirable) responses by other parts of the 'machine.'
3.3 Enzymes as drug targets
Enzymes are a major target for drugs. Their function is to catalyse reactions that take place in the body, e.g. the hydrolysis of an amide bond:
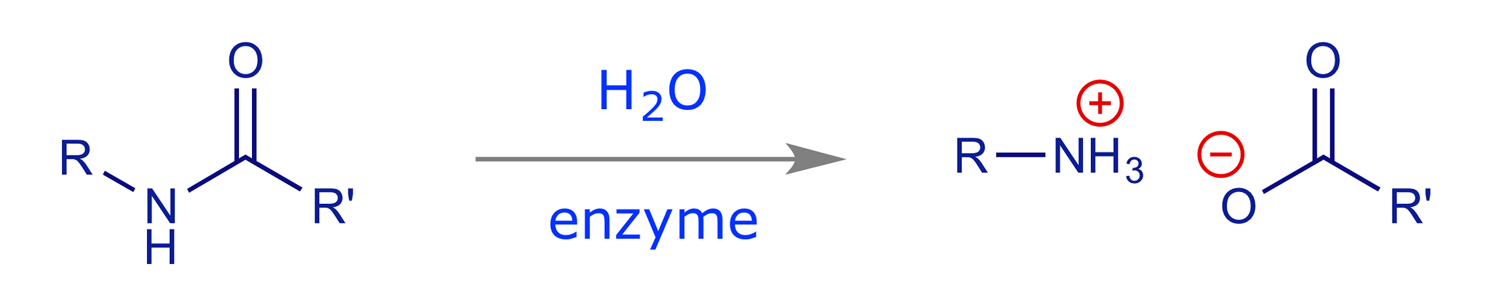
In the absence of the enzyme this reaction will occur, but is extremely slow (half-life many years). However, in the presence of the enzyme it is fast. Typical rate increases for enzyme-catalysed processes are 1010 to 1012, and typical turnover numbers are 103 molecules/min (i.e. one enzyme molecule will convert 1000 molecules of substrate into product every minute).
A rough scheme of how an enzyme works is as follows:
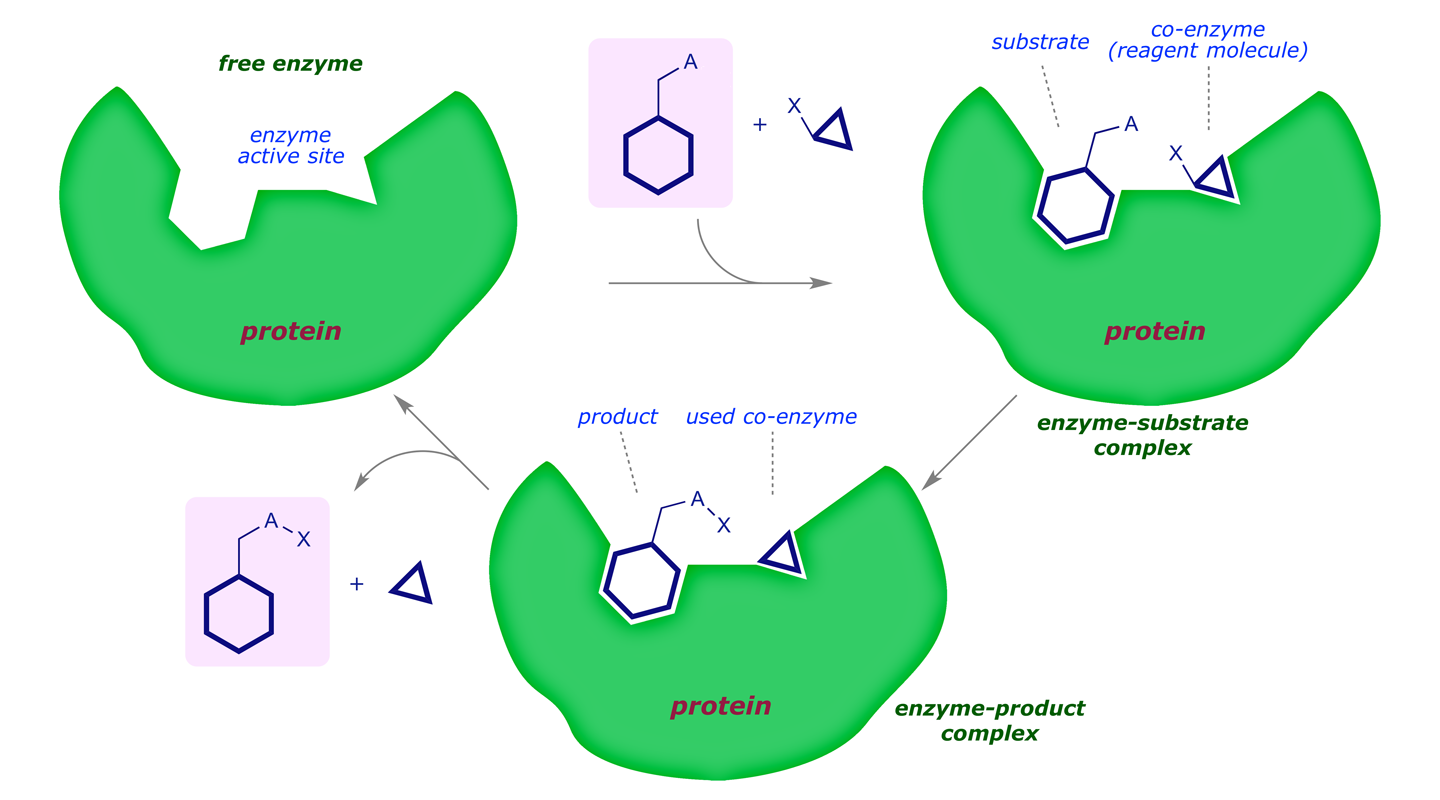
In energetic terms we see the following reaction profile:
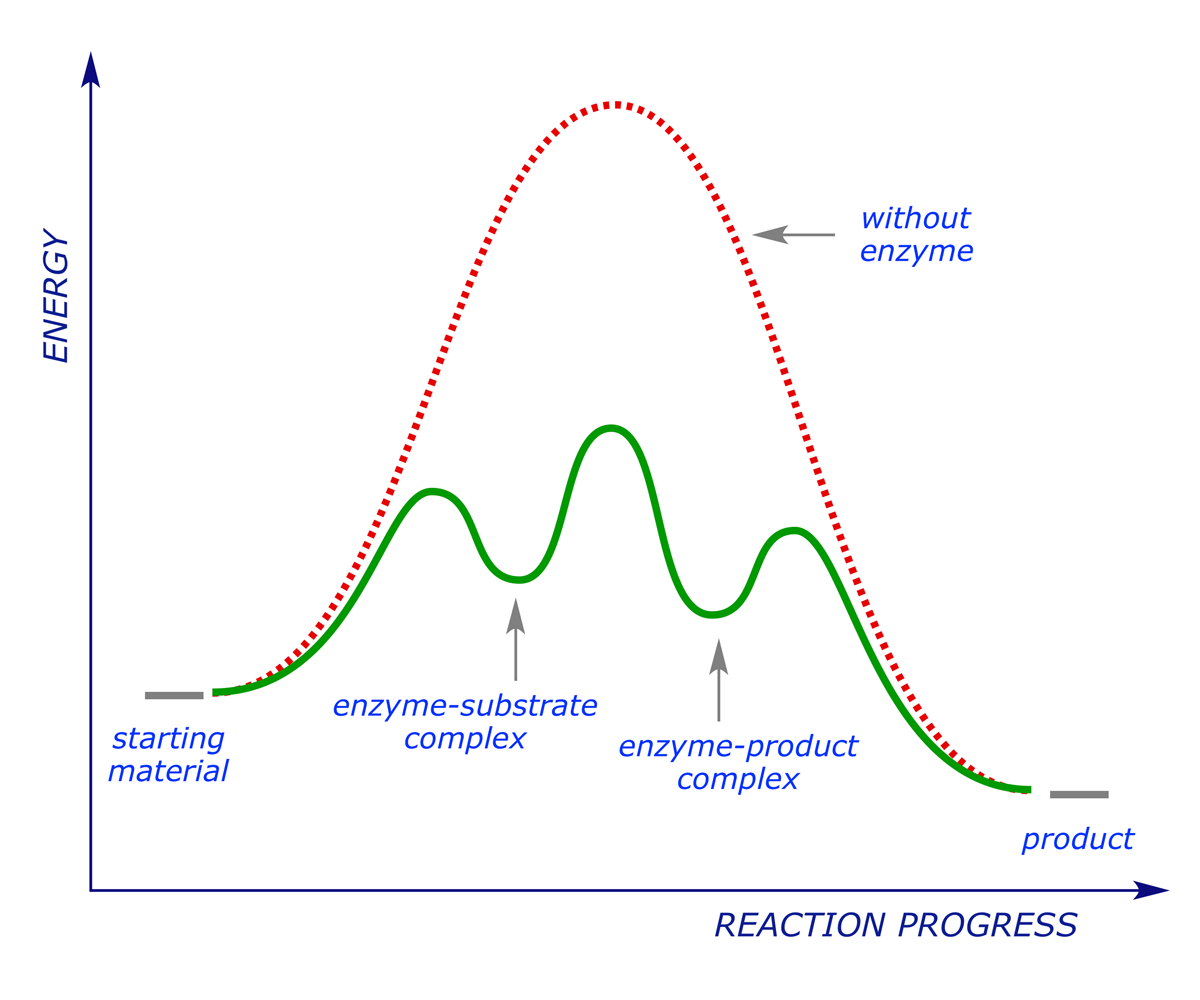
The enzyme works by stabilising the transition state for the reaction (i.e. reducing the activation energy). A drug that acts on an enzyme target blocks the active site, preventing the natural substrate from binding (and subsequently reacting). In this sense it operates in the same way as an antagonist acts on a receptor, but it should be noted that because enzymes work in a different way from receptors, it is not usually possible for a drug to behave as an agonist in this context — either the drug blocks the active site and the reaction is inhibited, or it does not block the active site and has no effect.
Because enzymes are chiral (built from 'handed' amino acids), they are usually stereoselective and will generally only operate on a small range of substrates. It is important to ensure that a drug only blocks the action of the target enzyme, and no others. The active sites of enzymes differ greatly, and as long as the differences can be exploited, this should be possible.