Sigmatropic Rearrangements
- Thermal [1,5] and [1,7] sigmatropic hydrogen shifts
- Thermal [1,3] sigmatropic hydrogen shift
- Photochemical [1,3] sigmatropic hydrogen shift
- Thermal [1,3] sigmatropic carbon shift
- [3,3] Sigmatropic rearrangements
In a sigmatropic reaction a σ bond 'migrates' across a conjugated system to a new site. If '[n,m] sigmatropic rearrangement' has n = 1, then one end of the bond that moves remains attached to its original atom — the n refers to the fact that the other end of the bond migrates across n atoms. This type of reaction often features a migrating C–H bond, i.e. a [1,n] hydrogen shift.
1 Thermal [1,5] and [1,7] sigmatropic hydrogen shifts
Example 020 features a suprafacial [1,5] shift. If we add one more C=C π bond (two more p electrons), the observed thermal [1,7] shift is antarafacial — the C–H starts 'above' but ends up 'below' and the H appears to migrate from one face of the π system to the other. This will only happen if the molecule can adopt the conformation required for the rearrangement.
![Thermal [1,5] suprafacial and [1,7] antarafacial sigmatropic H shifts](images_PR/6003.png)
As with electrocyclic reactions, we can use a simple FMO analysis in order to understand the selection rules for this type of reaction, but we will take a different approach this time. We will 'construct' the transition structure for the sigmatropic rearrangement and then apply the constraints of a pericyclic reaction — i.e. that the reaction is concerted and the transition structure must be a closed orbital loop. We will analyse in detail the thermal suprafacial [1,5] shift, and then generalise the results to other classes of sigmatropic rearrangements.
![A [1,5] sigmatropic H shift interpreted as a [π4s + σ2s] cycloaddition](images_PR/6006.png)
In the example shown above, the C–H is moving across ('reacting with') the π system, so we need to use either the HOMO of the C–H bond and the LUMO of the diene or the LUMO of the C–H bond and the HOMO of the diene [although it's easier to recognise the best overlap if we use the HOMO of C–H and LUMO of the diene]. As expected, the result is the same whichever pairing we choose — we find that a suprafacial [1,5]-sigmatropic rearrangement is thermally allowed. The transition structure for such processes is relatively easy to reach and such reactions are common. Thermal [1,7] hydrogen shifts are antarafacial, and many examples of this process are known.
The graphic below shows how the orbital 'matching' is once again switched by the addition of an extra π bond. The terminal lobes of the diene LUMO are in phase, and match those of the σ bond HOMO, making the 'cycloaddition' of the diene to the σ bond a thermally allowed suprafacial + suprafacial process. This is not the case with either the alkene or the triene.
![The LUMO of the π-system is used to predict the outcome of thermal [1,n] H shifts](images_PR/6009.png)


2 Thermal [1,3] sigmatropic hydrogen shift
This reaction is not observed, and we can see why when we perform the usual analysis (Figure 6.3). The reaction cannot take place suprafacially while maintaining bonding interaction between the H and the ends of the π system. Thermal suprafacial [1,3]-sigmatropic H shifts are forbidden.
![A thermal [1,3] antarafacial H shift, which is 'allowed' but physically impossible](images_PR/6018.png)
If the H atom were able to move from 'above' to 'below' as illustrated by the blue dashed line in Figure 6.3, then the required bonding interactions would be maintained in the transition structure. However, it is physically impossible for the H to achieve this while maintaining bonding overlap throughout the required cyclic transition structure. In consequence, a thermal antarafacial [1,3]-sigmatropic rearrangement is allowed electronically but in practice it is prohibited by the structural demands of the mechanism.
3 Photochemical [1,3] sigmatropic hydrogen shift
This reaction is observed, and again we can analyse the FMOs to see how it works. After UV irradiation the HOMO of the alkene bond is π*, and in order to effect our 'cycloaddition' type of analysis we must combine this with the LUMO of the C–H bond.
We get the same result if we view the reaction as the cycloaddition of a proton to an allyl anion, choosing the LUMO of H+ (little choice here) and the HOMO of allyl anion (ψ3 in the photochemically excited state).
![A [1,3] suprafacial H shift shown as the cycloaddition of a proton to an allyl anion](images_PR/6024.png)
![A photochemical [1,3] suprafacial H shift, which is physically easy](images_PR/6021.png)
The reaction can take place suprafacially and maintain simultaneous bonding interactions between the H and the ends of the π system. A photochemical suprafacial [1,3]-sigmatropic rearrangement is allowed (and of course, being suprafacial, is physically possible).
We can now see that the selection rules emerging from the above analysis are based, as with other pericyclic processes, on the number of electrons in the transition structure (which equals 2 x the number of curved arrows in the mechanism):

4 Thermal [1,3] sigmatropic carbon shift
In Section 2 we saw that a thermal [1,3] hydrogen shift is forbidden because bonding overlap at both ends of the π system in the transition structure is impossible. The thermal [1,3] shift of a saturated carbon by mechanism A (below) is precluded for the same reason: The reaction cannot take place suprafacially at the migrating carbon with simultaneous bonding between the C atom and the two ends of the π system, which must react antarafacially.
![A thermal [1,3] C shift, with the C reacting suprafacially, is 'allowed' but physically impossible](images_PR/6030.png)
However, an alternative reaction pathway is available to carbon by virtue of the rear lobe of the sp3 hybrid orbital of the participating C–C σ bond. This allows the migrating carbon to serve as the antarafacial component that this type of reaction requires (no such pathway is open to hydrogen because its bonding orbital has only a single phase).
![A thermal [1,3] C shift, with the C reacting antarafacially, is possible](images_PR/6033.png)
The alternative pathway (B) requires the inversion of configuration at the migrating carbon atom, and this prompted researchers to design experiments that would reveal its existence. The example 040 below, published by Berson and Nelson in 1967, provides a remarkable demonstration that, albeit under harsh thermal conditions, the atoms C(3) and C(7) can engage in the bonding overlap (dashed line labelled a) required to bring about the 1,3-shift of the C(1)–C(7) bond via a formal [π2s + σ2a] process. The Berson paper is worth reading to learn how the starting materials were made, as well as to see how the results were verified.
![The thermal [1,3] C shift described by Berson and Nelson (1967)](images_PR/6036.png)
![Orbitals involved in the thermal [1,3] C shift reported by Berson and Nelson](images_PR/6039.png)
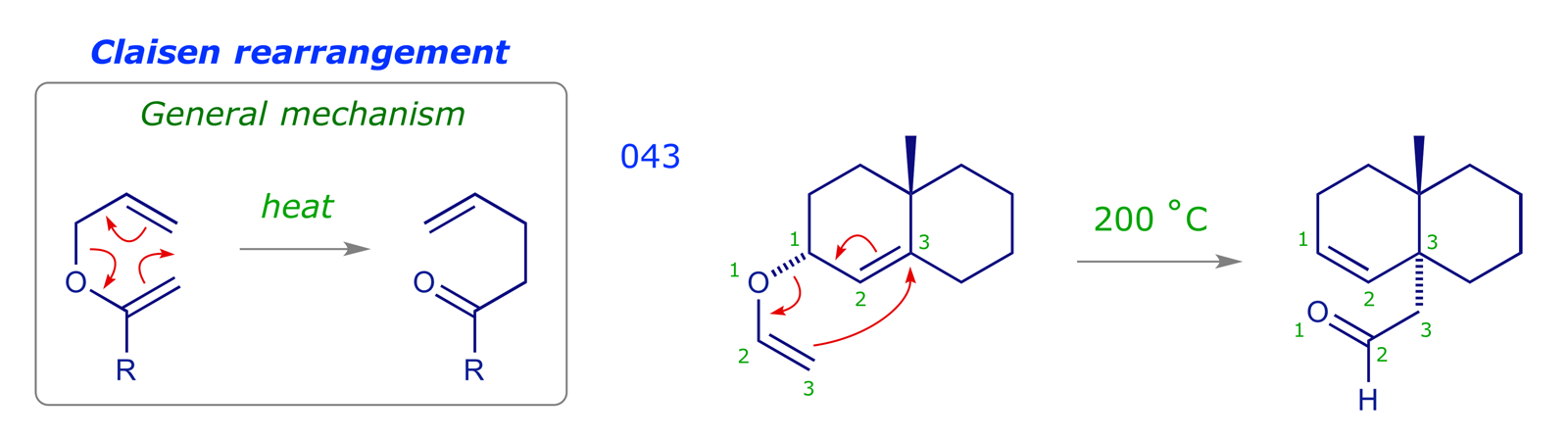
5 [3,3] Sigmatropic rearrangements
The other class of pericyclic reactions we will cover is [3,3] sigmatropic rearrangements. In these, both ends of the relocating σ bond migrate three atoms.
![The numbering system used for a [3,3] sigmatropic rearrangement](images_PR/6042.png)
There are two important general classes of these reactions. The 'all carbon' version is known as the Cope rearrangement, and an oxygen in the appropriate position changes this to the Claisen rearrangement. There is a series of structural variants based on these two reactions.

5.1 The Cope rearrangement
In order to construct the transition structure we will require two 3-carbon units: These are the allyl anion (HOMO) and the allyl cation (LUMO). In fact these are the same orbital, and combining them shows that both components can react suprafacially in a thermal process. It is reasonable to predict a chair-like transition structure structure (compare with aldol addition).
![The Cope rearrangement analysed as the [4 + 2] cycloaddition of allyl anion to allyl cation](images_PR/6051.png)
Cope rearrangements can be driven by the relief of strain, as illustrated by the following synthesis of the seaweed pheromone, dictyopterene C.

The chair-like transition structure, illustrated above, is similar to the Zimmerman-Traxler transition structure for the aldol addition reaction, and has analogous stereochemical consequences for the Cope rearrangement. If a substrate carries a substituent, there will be two possible TS conformations, with the substituent taking either an axial or an equatorial orientation. In the lower energy of these the substituent will be equatorial.
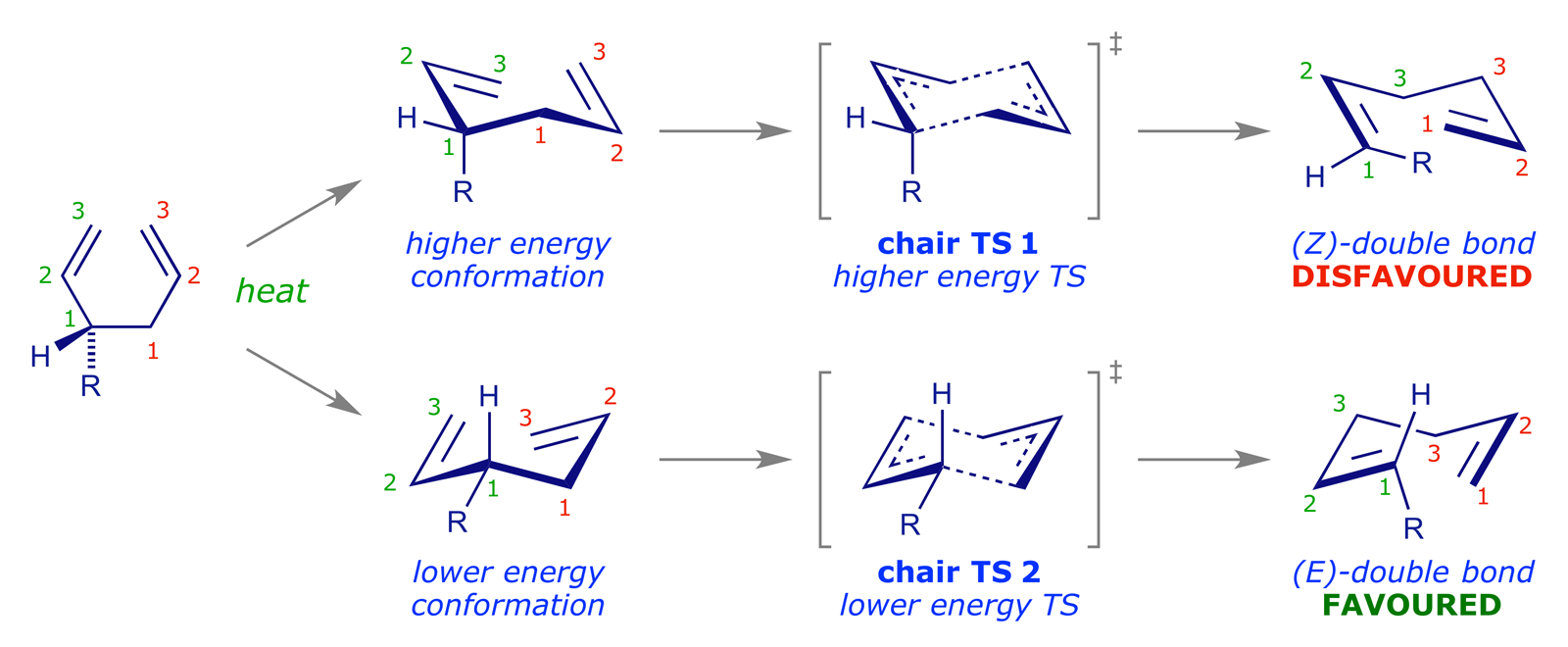
The following examples show this stereochemical preference in action.
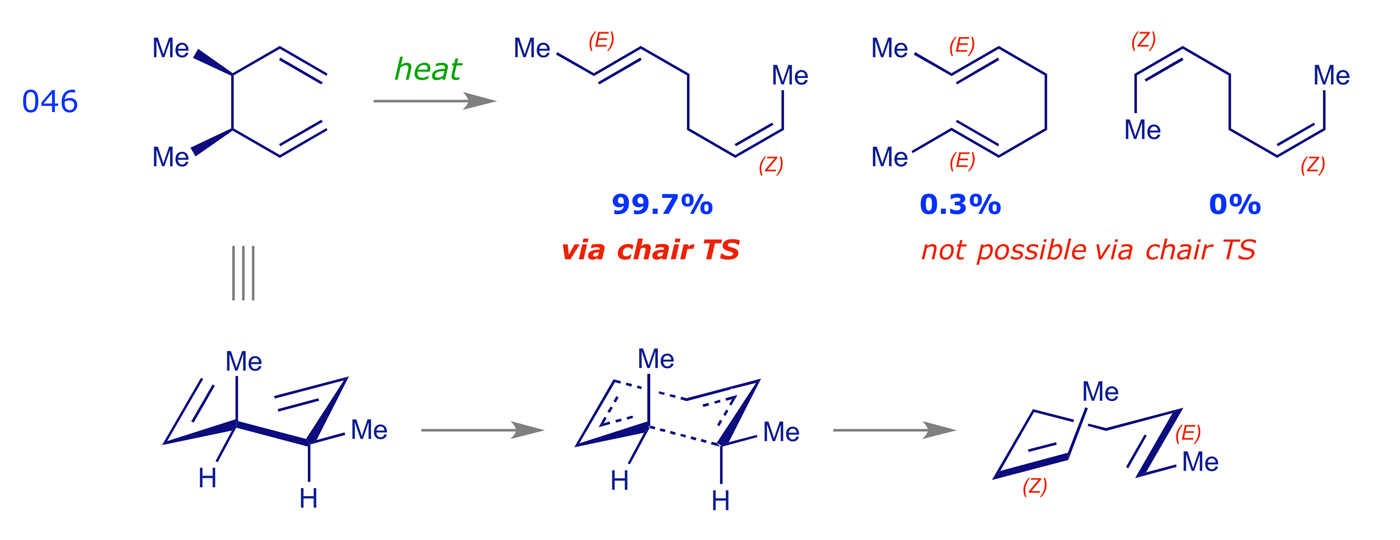

Cope rearrangements are reversible, the equilibrium lying in favour of the more stable species. In the examples 042, 046 and 047 the driving force is the formation of double bonds that are more substituted (trisubstituted are more stable than disubstituted, etc.). The dictyopterene C' synthesis 045 is driven by the relief of cyclopropyl ring strain. Example 009 illustrates an oxy-Cope rearrangement, in which the pericyclic reaction results in the formation of a strained 10-membered ring; the subsequent enol-keto tautomerisation step provides a driving force for the overall process.
![[3,3] Sigmatropic (oxy-Cope) rearrangement of 1,2-divinylcyclohexanol](images_PR/6066.png)
5.2 The Claisen rearrangement and variants
If we change the appropriate carbon in a Cope rearrangement for an oxygen we arrive at the Claisen rearrangement, the general mechanism of which is shown below.

- [3,3]-Sigmatropic rearrangement
- Thermally allowed
- Carbonyl product about 84 kJ/mol (20 kcal/mol) more stable than the starting enol ether
- Chair-like transition structure, predictably stereoselective
- Variants differ only in the substituent Y
The existence of so many variants of a reaction usually means that (a) the reaction is very useful (otherwise there's not much point in variants) and (b) there is a problem with the original version. In practice there are two issues that limit its usefulness; firstly, high temperatures can be necessary; secondly, it can be difficult to prepare the required alkenyl ether (normally not a problem with R2 = H, i.e. vinyl ethers, which can be made in a simple exchange process using ethoxyethene).
The enolate-Claisen is the simplest variant. It requires only the preparation of an ester, which is treated with strong base to form the enolate. This is then allowed to warm up. Problems can be encountered with this method as some enolates can decompose before rearrangement. However, the reaction can be very useful in some cases (see example 044).


For the Ireland-Claisen, an ester enolate is prepared as above, but before allowing it to warm up the enolate is O-silylated with a chlorosilane (R3SiCl). The resulting silyl enol ether (also known as an 'enol silane') is much more stable than the enolate. On heating it does not decompose, but smoothly undergoes the rearrangement.

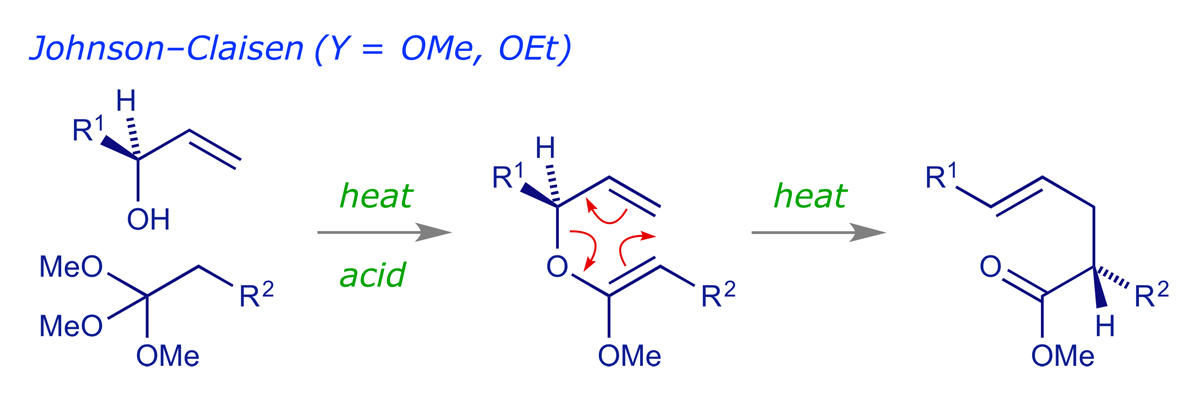
In the Johnson-Claisen rearrangement, the starting material is prepared by the acid-catalysed exchange of an orthoester (1,1,1-trialkoxyalkane) with an allylic alcohol, followed by elimination to give the ketene acetal, which undergoes rearrangement as it is formed.